
記住我










Cystic fibrosis (CF (OMIM 219700)) is characterised by a triad of chronic obstructive pulmonary disease, exocrine pancreatic insufficiency and elevation of sodium and chloride concentration in sweat caused by biallelic pathogenic variants in the CFTR gene.1 2 The disorder was first described in 19383 and was originally known as mucoviscidosis, given the observation that the patients presented abnormally thick mucus (reviewed in a previous work4).
Mucus is an essential polymer secreted mainly by specialised cells in the respiratory and digestive tracts. It plays a vital role in the protection against infectious and toxic agents by clearing debris and pathogens through mucociliary clearance. Furthermore, mucus also protects the sensitive epithelial surfaces in the airways and intestine.5 Mucins are the major macromolecular constituents of epithelia mucus and play a relevant role in health and disease.6 The secreted or gel-forming mucins are responsible for the viscoelasticity of mucus, with MUC5B and MUC5AC being the major gel-forming mucins present in the airways, where they have different airway clearance functionalities.7 8 In the small intestine and colon, MUC2 forms the layer of mucus and is responsible for the protection of the gut barrier, the regulation of microbiome homeostasis and the prevention of diseases.9 10
Within this study, we present 13 patients from 9 unrelated families suffering from a previously undescribed genetic disorder characterised by recurrent lower respiratory infections, chronic diarrhoea and failure to thrive—a phenotype clinically resembling cystic fibrosis (CF). By performing exome/genome sequencing (ES/GS) and an analysis of our rare disease-centric Bio/Databank, we identified six different AGR2 biallelic variants as disease-causing for these patients. In mice, Agr2 is relevant for the normal functioning of mucins from the respiratory and gastrointestinal tract,11–13 suggesting that AGR2-related disease might be a novel mucus disorder.
MethodsThis project has been conducted within a diagnostic setting, and as a second step, used deidentified data and samples. Thus, this did not require institutional review board (IRB) approval in our jurisdiction. Written informed consent was obtained from all nine families for genetic studies as well as for scientific publication of anonymised clinical data and clinical photographs. Additionally, the consent declaration included information regarding storage of the data and further processing for research purposes. The informed consent form is available in English and several other languages (https://wwwcentogenecom/downloadshtml).
Exome sequencing (ES)DNA was extracted using standard methods from dried blood spots (DBS) submitted on filter cards (CentoCard®). Details of the laboratory procedures, bioinformatics analysis and evaluation of the exome data are provided in the Supplemental data. AGR2 exon numbering was based on transcript NM_006408.3 (8 exons). Variant nomenclature followed standard Human Genome Variation Society recommendations.
Analysis of proprietary Bio/DatabankOur Bio/Databank14 contains data from 65 005 individuals with ES and/or GS data, as well as corresponding clinical information, which is registered in the Bio/Databank as human phenotype ontology (HPO) terms. A total of 39 756 of these individuals are patients with at least one HPO term used for phenotype description. After identification of AGR2 as a candidate gene in the first family, the Bio/Databank was investigated for rare biallelic variants in the AGR2 gene (ExAC/gnomAD<1%). Variants with high or moderate predicted impact on protein structure or function (CADD raw score ≥4) were prioritised. For the identified cases, HPOs, all available clinical information, and test results were reviewed to elucidate the associated phenotype. The ES and/or GS data were reanalysed to investigate whether other variants could contribute to the phenotype of the patients. Referring clinicians were then recontacted (for cases with consent provided).
RNA sequencing and data analysisNasal swabs were taken for the probands (families 2, 3, 4, 6, 7 and 8) using ORE-100 RNA collection kits (Steinbrenner Laborsysteme GmbH), and RNA was extracted with the Quick-RNA micro prep kit (Zymogen) following manufacturer’s instructions. The TruSeq stranded mRNA kit (Illumina) was used to generate next-generation sequencing (NGS) barcoded libraries. After pooling, the libraries were sequenced on a NextSeq 500 system using the 75 bp paired-end protocol. An average of 34 052 907 reads (>Q30) were obtained for each sample. RNA-seq reads were aligned using two-pass mode with STAR V.2.7.6a15 against human genome GRCh37/Release 38 (www.gencodegenes.org). The read groups were fixed, and the duplicates were marked using Picard tools V.2.23.8. Counting the reads was performed by featureCounts/subread V.2.0.1.16 Initial quality control and differential expression analysis was performed with DESeq2_1.32.0,17 and pathway analysis was performed with ToppGene.18
Total RNA was converted to cDNA using reverse transcriptase Superscript IV (Invitrogen). Primers were designed to amplify AGR2 fragments from exons 1–8, 2–6 and 4–7 (primer sequences available on request). After PCR, aliquots were electrophoresed on 1% agarose gels at 90 V for 90 min, stained with SYBR safe (Invitrogen).
Protein structural analysisExperimentally solved structures for both the monomeric and dimeric conformations of AGR2 can be found in the Protein Data Bank. These files, PDB-codes 2LNT and 2LNS, respectively, contain the C-terminal residues 36–175. We used the YASARA19 and WHAT IF software20 to study these structures.
Ceramide26 quantification in DBSC26 Ceramide species were quantified in DBS extracts using a method previously described,21 as well as multiple reaction monitoring mass spectrometry.
A detailed methods description can be found in the Supplemental data.
ResultsClinical description of the affected individualsThirteen patients from nine families presented with a similar CF-like phenotype consisting of recurrent respiratory infections, chronic diarrhoea and failure to thrive (online supplemental table 1). The patients’ ages ranged from 10 months to 10 years old. Symptoms started usually around the neonatal period. The children suffered from recurrent coughing, wheezy episodes, pneumonia, interstitial lung disease and bronchiectasis. Further episodes of vomiting and chronic diarrhoea led to poor weight gain. Additionally, four patients presented hepatosplenomegaly, and two had cardiovascular abnormalities (mitral valve insufficiency and right heart failure with severe pulmonary hypertension). Most patients had appropriate neurodevelopment; only two cases presented developmental issues. Respiratory and gastrointestinal complaints caused frequent and prolonged hospital admissions. Family history was positive in four families (out of eight) with several similarly affected relatives. Sweat chloride tests and pancreatic elastase results were normal (when performed). Two patients had cultures positive for Pseudomonas. Patient III-1 (family 6) had a bronchoalveolar lavage cytology that showed cellular fluid composed of bronchial epithelial cells and alveolar macrophages with strands of thick mucus. Nasal ciliary brush study was done in two patients, with motile cilia seen under light microscopy with 9+2 normal configuration (family 6 and family 7). However, ultrastructural electron microscopy analysis of a nasal brush sample (family 8) detected ciliary abnormalities in 34% of the 189 examined transverse cilia sections. The abnormalities were related to missing central doubles, triplets instead of central doubles with missing dynein arms (inner and/or outer) at peripheral doubles, and duplication of central doublets with missing dynein arms (inner and/or outer) at peripheral doublets (online supplemental figure 1). This result was considered as inconclusive since in patients with a primary ciliary defect, most cilia would be expected to be abnormal.22
Additional clinical data can be found in the supplementary data (online supplemental table 1 and Clinical summaries).
Exome and Bio/Databank analysesIn family 1, exome analysis focused on ‘diagnostic’ genes which did not detect any relevant variant. We then performed an extended exome analysis focusing on genes not yet linked to a human phenotype. Given the positive family history and parental consanguinity, we prioritised homozygous variants. Eight variants were identified (online supplemental table 2), from which the novel missense variant in the AGR2 gene (NM_006408.3:c.211C>A, p.Pro71Thr) was selected as the best candidate given its rarity, high conservation, and known function and protein localization.12 13 23 Targeted testing confirmed that the parents were heterozygote carriers, and both affected cousins were homozygotes for the same variant, fully co-segregating with the disease (figure 1).
 Figure 1
Figure 1 Summarised family trees of the nine families and the identified AGR2 variants. Variants are colour-coded, the founder missense variant is shown in red font, with the corresponding haplotypes (families 2, 3, 4 and 7). Genotypes are shown below available individuals. AGR2 genotypes show full co-segregation with the phenotype.
This finding prompted us to mine our Bio/Databank to identify additional cases with overlapping phenotypes, no genetic diagnosis established, and rare biallelic variants in the AGR2 gene. Exome/genome data from 39 756 patients was investigated. We prioritised AGR2 rare, homozygous and compound heterozygous variants, with predicted impact on the protein. Additionally, we searched for copy number variants (CNVs) affecting the AGR2 gene.
This resulted in the identification of a total of 13 patients from nine families (figure 1 and online supplemental table 1). All patients presented with very similar phenotypic features, mainly including recurrent lower respiratory tract infections, and no evident immunological abnormalities. Relevant rare homozygous variants are shown in online supplemental table 2. The missense variant in exon 6 of AGR2 (NM_006408.3:c.349C>T, p.His117Tyr) was present in patients from families 2, 3, 4 and 7. Analysis of the genomic region around AGR2 indicated a shared haplotype for these families (from rs71523042 to rs780638101, 2.8 Mb), suggesting a common ancestor. Families 2, 4 and 7, from Syrian origin, had a larger shared chromosomal region of approximately 8.2 Mb (figure 1). An additional missense variant was identified in family 9 (c.428G>A, p.Gly143Glu).
We also detected two splice site variants (NM_006408.3:c.330+1G>T and c.330+1del) in families 5 and 8, respectively. The variant was confirmed as heterozygous in the parents and unaffected sibling and homozygous in the similarly affected brother in family 5 (figure 1). The index from family 8 is adopted, and no biological relatives could be tested. The variants affect the canonical splicing site and are predicted to abolish normal exon 5 splicing.
Furthermore, we identified a large homozygous deletion including exon 1 to exon 7 of the AGR2 gene and affecting the neighbouring gene AGR3 in family 6 (full gene deletion, genomic coordinates chr7:16,834,229–16,936,407, figure 2A). This prompted us to query our Bio/Databank for potential causative variants in AGR3. We identified several individuals with homozygous missense, splicing and nonsense variants in AGR3. However, these variants were present in unaffected adults (parents) and patients with variable phenotypes not overlapping with the clinical features of the patients described in this study. Therefore, these data do not support a causative effect of AGR3 variants.
 Figure 2
Figure 2 AGR2 variants identified in the patients and abnormal splicing caused by an intronic variant. (A) The deletion region of AGR2/3 is shown in the Integrative Genome Viewer (IGV). Reads for the exonic regions of the AGR2/3 genes can be seen in the control (lower panel), whereas no reads are seen in the index sample III-1 (deleted region is boxed, chr7:16834456–16918247). This deletion was confirmed by qPCR. (B) Schematic representation of AGR2 gene, with the detected variants shown (font colours match the respective family). (C) Sashimi plots from IGV, illustrating AGR2 splicing junctions. Arcs represents splice junctions and connect the exons, the number of reads split is displayed across the junction. The variant c.330+1del causes aberrant splicing, note the junctions skipping exon 5 (arrow). See also online supplemental figure 2A–C.
We also searched other data repositories for variants, such as gnomAD (v2.1.1) and Decipher for AGR2 variants (SNVs and CNVs). Genes related to autosomal recessive diseases are relatively unconstrained.24 However, loss of function (LoF) variants in AGR2 are ultrarare, with not a single individual reported as homozygote in these data repositories. The variants detected in our patients are novel or ultrarare in gnomAD, with high conservation and CADD scores supporting adverse consequences (online supplemental table 3).
RNA sequencing analysisThe AGR2 protein is mainly detected in mucus-secreting organs from the gastrointestinal tract, the respiratory tract and the reproductive system.23 To assess the effect of the splicing variant and the large deletion and to learn about the putative affected pathways, we performed RNA sequencing using RNA isolated from the nasal mucosa from 6 patients, 4 heterozygote carriers and 11 controls. RNA sequencing confirmed that the c.330+1del variant causes aberrant splicing of AGR2, disturbing exon 5 splicing with retention of intronic regions and altering the reading frame, finally leading to LoF (figure 2C and online supplemental figure 2AC). Abnormal splicing was confirmed by targeted AGR2 RT-PCR (online supplemental figure 2B). We also confirmed that the large deletion detected in the index patient from family 6 leads to a complete loss of AGR2 transcripts (online supplemental figure 2C). Both patients had very low to nearly no AGR2 expression (adjusted p value=0.002), confirming that both variants are leading to LoF. For the founder missense variant c.349C>T, there is no evident splicing effect (figure 2C and online supplemental figure 2A-C). Additionally, differential gene expression analysis detected biological processes that were significantly dysregulated in the patients compared with control and carrier samples. Processes such as cell/leucocyte activation and others related to the immune system were transcriptionally upregulated, whereas processes such as microtubule-based movement, process, transport and cilium organisation were significantly downregulated (online supplemental file 1). As an exploratory analysis, we evaluated differential gene expression in the two patients with proven LoF variants (large gene deletion and c.330+1del) compared with the controls. Two relevant mucins (MUC2 and MUC5AC) and CLCA2 (from the calcium-dependent chloride channel family) are at the top of the downregulated genes (online supplemental file 1).
Protein structural analysis (missense variants)Based on the published AGR2 protein structure,25 we investigated the possible effects of the missense variants p.Pro71Thr, p.His117Tyr and p.Gly143Glu. We first examined whether the variants could directly affect dimer formation. The Pro71 residue is semi-buried in the core of the protein, whereas the His117 and Gly143 occur in surface loops but are not immediately at the dimerisation face (figure 3A and B). For Pro71Thr, the proline side chain is slightly larger than threonine; the main differences between these two residues lie in the tendency of the proline residue to make rigid turns that stabilise the protein structure (figure 3C). A substitution of histidine by tyrosine would result in loss of an amino acid that can potentially store electrons (figure 3D). Lastly, Gly143Glu is a clear example of the introduction of a larger side chain that will no longer fit at that position. Glutamic acid with its charged γ-carboxyl side chain would be disruptive and large compared with the glycine with its single hydrogen side chain (figure 3E). This change will affect the local structure simply by restructuring the surrounding residues to remove steric clashes, and may affect interactions with other proteins by changing the surface of AGR2.
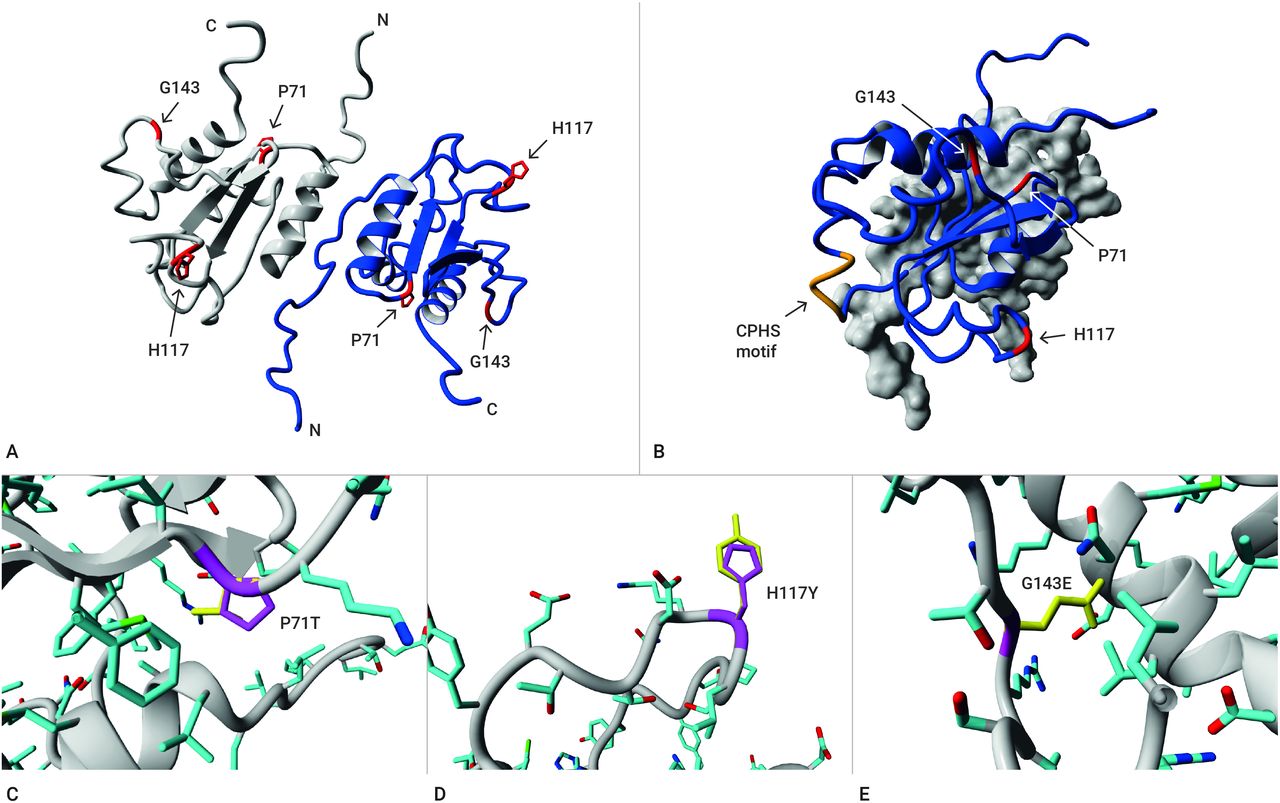 Figure 3
Figure 3 Structural protein analysis of the missense variants. (A) Dimer of the AGR2 residues 36–175. Monomers are individually coloured in grey or blue. Side chains of the residues are not shown, except for the mutated P71, H117 and G143 in red. (B) Overview of AGR2 as seen from the side; one monomer is shown as grey surface only. This view shows the distance between the mutated residues (red side chains) and the putative active site of the protein CPHS-motif (orange). (C) Variant P71T: The proline side chain is shown in magenta; note the attachment of the side chain to its own backbone; the threonine side chain is shown in yellow. Side chains of the protein are coloured by atom type (carbon=cyan, oxygen=red, nitrogen=blue, sulfur=green). The proline side chain is slightly larger than threonine, but the main differences between these two residues lie in the shape of the side chain and proline tendency to make rigid turns that stabilise the protein structure. (D) Variant H117Y: The histidine side chain is shown in magenta, whereas the tyrosine side chain is yellow. Other atoms are coloured as described. The change from histidine to tyrosine indicates a small difference in size, and a different potential for interactions since histidine’s side chain can be used for electron storage. (E) Variant G143E: The side chain of the mutant residue glutamic acid will not fit in the same space (note that wild-type glycine does not have a side chain). The change in charge and side chain size will affect the local structure and may affect interactions with other proteins.
Whole blood ceramides (Cer26) analysisLipid metabolism imbalances have been consistently reported in patients with CF, as measured in plasma,26 human primary bronchial cells27 and mesenchymal stem cells.28 Among sphingolipids, ceramide is emerging as one of the players of the pulmonary dysfunction in inflammatory lung diseases; enhanced sphingolipid metabolism leads to an increased ceramide content, which in turn contributes to maintaining the chronic inflammatory status.28 We measured the levels of ceramide26 (Cer26) in DBS from patients with homozygous biallelic variants in AGR2 (n=5), in molecularly confirmed patients with CF (n=11) and healthy controls (n=10). Patients with CF had slightly lower Cer26 levels, but this difference was not significant. All four AGR2 patients with severe pulmonary disease consistently showed pathologically elevated levels of Cer26cis, Cer26trans and Cer26total isomers, while the patient with mild respiratory symptoms showed normal levels (family 3). Taken as a whole, the AGR2 patient group had significantly elevated levels of all measured Cer26 isomers when compared with patients with CF and healthy controls (Cer26total F=10.94, p<0.001; online supplemental figure 3). These findings suggest an altered Cer26 metabolism in patients with AGR2-related disease.
DiscussionBy combining ES and Bio/Databank analyses, we identified 13 patients from 9 families with rare homozygous variants in AGR2 (figures 1 and 2). Three of these variants are very likely leading to a LoF (affecting canonical splicing site and large gene deletion), suggesting that loss of AGR2 is likely causing the phenotype in at least a subset of these patients. Affected individuals presented very early in life with recurrent coughing, wheezing, low tract respiratory infections, chronic diarrhoea and failure to thrive which resembled CF (online supplemental table 1). However, these patients presented normal sweat/elastase tests. Although four of them presented hepatomegaly with undetermined cause, meconium ileus, pancreatic insufficiency, steatorrhea or pancreatitis were not reported in our patients. Thus, on a closer look, CF can be clinically excluded in these patients. This was later confirmed by genetic testing with no relevant variants detected in the CFTR gene. From a clinical perspective, it is important to consider AGR2-related disease as a differential diagnosis of patients presenting a CF-like phenotype (and normal sweat/elastase tests).
Interestingly, the index case from family 8 presented cilia abnormalities in the nasal epithelium (online supplemental figure 1), although these could be secondary cilia changes, as described also in CF and chronic bronchitis.29 Together with our results from the RNA differential expression and pathway analysis, cilia abnormalities may occur as part of the AGR2-related phenotype; however, more patients would need to be examined.
The AGR2 protein is detected at high levels in tissues that secrete mucus or function as endocrine organs, including the respiratory tract, stomach, colon, prostate and small intestine (reviewed in a previous work23). At the cellular level, AGR2 is expressed in Paneth and goblet cells (intestine/colon), ciliated cells (airways) and glandular cells (pancreas and prostate, among others)12 30 31 (Human Protein Atlas, http://www.proteinatlas.org). Subcellularly, AGR2 localises to the lumen of the endoplasmic reticulum (ER), indirectly associates with ER membrane-bound ribosomes, and it is involved in the maintenance of ER homeostasis. Knockdown of AGR2 significantly alters the expression of components of the ER-associated degradation machinery and reduces the ability of cells to cope with acute ER stress.32
AGR2 is required for adequate production of intestinal mucin MUC2 and airway mucins MUC5B and MUC5AC.11–13 33 Mouse Muc5b is required for mucociliary clearance, for controlling infections in the airways and middle ear, and for maintaining immune homeostasis in mouse lungs, whereas Muc5ac is dispensable.34 On the other hand, MUC2 is the major intestinal mucin,11 35 and it has been implicated in inflammatory bowel disease and colorectal cancer.36 37 Agr2 knockout mice are born healthy but are unable to produce intestinal mucin and are highly susceptible to experimentally induced colitis, with profound weight loss and intestinal bleeding suggesting a role of Agr2 in protection from disease. With ageing, Agr2 knockout mice develop rectal prolapse, a feature observed in mouse models with colitis.11 Extensive spontaneous ileitis and colitis were also described in mice lacking Agr2.12 Schroeder et al described a considerable reduction of Muc5ac and Muc5b in the airways of allergen challenged Agr2-deficient mice, with abnormal allergen response compared with wild-type controls. This is likely due to impaired mucin transit through the ER, where these mucins were found to accumulate.13
Thus, the evidence presented in the studies of the Agr2-deficient mice points to an important role of Agr2 in mucin/mucus production, as well as homeostasis of the respiratory and intestinal tract. These findings are also compatible with the phenotype observed in the patients described in this study and support the hypothesis that the detected variants are likely acting via a LoF mechanism.
Interestingly, one patient presented a large homozygous deletion affecting seven out of eight exons of AGR2 and the complete AGR3 gene. The Agr3 protein is detected in ciliated cells in the airway epithelium, and unlike Agr2, it is not induced by ER stress. Mice lacking Agr3 are viable and develop ciliated cells with normal-appearing cilia, which have reduced ciliary beat frequency in the airways, associated with impaired mucociliary clearance in Agr3-deficient animals.38 No differences in phenotype were observed between the patient with the homozygous AGR2-AGR3 deletion and the rest of the patients. Cases with AGR3 biallelic variants and overlapping phenotypes have not been described, to our knowledge.
Pathway transcriptome analysis of nasal samples from patients, carriers and healthy controls detected upregulated biological processes such as immune response, leucocyte activation and immune effector processes which could be related to the recurrent airway infections suffered by the patients. Further, downregulation of cilia-related processes (intraciliary transport, microtubule-based transport, cilium organisation) could also reflect a defective cilia function in the patients. This could be secondary to a primary mucus abnormality.
Interestingly, a recent article reports two siblings with severe congenital enteropathy, but also recurrent respiratory infections and wheezing—a phenotype that overlaps with the clinical features of our patients. The siblings had the same homozygous missense founder variant (c.349C>T, p.His117Tyr) reported in five of our patients. The authors detected very low levels of MUC2 protein in the intestinal wall of the patients.30 They found high levels of mislocalised AGR2 protein in the epithelial surfaces of gastric and bowel sections of the patients compared with controls. In our transcriptome analysis, no significant differences in mRNA AGR2 expression were detected when comparing average AGR2 expression in patients, with carriers, or controls. Combining this information, this suggests that the mutant AGR2 (His117Tyr) protein is produced but has impaired functionality, probably leading to accumulation and mislocalisation at the affected epithelia. Since AGR2 is essential for MUC2/mucus production,11 loss of AGR2 functioning could explain the low levels of MUC2 protein detected intestinal mucosa of the patients compared with controls.30 This also aligns with our transcriptome differential gene expression analysis focused on the patients with proven LoF variants, which showed significantly reduced levels of AGR2, but also MUC2 and MUC2AC. Interestingly, this analysis also showed a significant reduction of CLCA2 levels. CLCA2 belongs to the calcium-dependent chloride channel family which are involved in the regulation of electrolytic fluxes and modulate secretion, absorption, cell volume and membrane potential, predominantly expressed in the digestive tract and trachea.39
Based on the AGR2 structure published by Patel et al,25 our structural analysis suggests that the missense variants detected in the patients could affect proper AGR2 interactions with other proteins (His117Thr) or might affect the protein structure (Pro71Thr and Gly143Glu). Importantly, His117Thr occurs roughly on the same side of the protein as Cys81. Change of Cys81 into serine is described as causing loss of interaction with Muc2.11 This is also in line with a recent cellular model that found reduced binding of mutant AGR2 (His117Thr) to MUC2.30
Sphingolipid metabolism and ceramide content is altered in airway epithelial cells from patients with CF.27 28 Ceramides are implicated in inflammation and their accumulation in CF cells was previously demonstrated.40 In Cftr-deficient mice, ceramide accumulation leads to constitutive age-dependent pulmonary inflammation, death of respiratory epithelial cells, deposits of DNA in bronchi and high susceptibility to severe Pseudomonas infections.40 We directly measured DBS extracts as validated by us previously.21 In four out of the five AGR2 patients, ceramide isomers (Cer26) were significantly higher than in healthy controls and patients with CF. Ceramides have been found consistently elevated in the airways of patients with CF and CF animal models, and its accumulation significantly contributes to sustained inflammation and inability to fight lung infections. Conversely, low plasma ceramides have been found in patients with CF, which has been attributed to the abnormal lipid metabolism and malabsorption (reviewed in a previous work41). We also detected lower Cer26 levels in patients with CF, although this difference was not significant. Our findings in AGR2 patients point to a role of ceramides, specifically Cer26 in the AGR2-disease pathophysiology. Whether this is part of a specific disease mechanism or a reflection of a systemic inflammation process, will require further investigation. Cer26 determination could potentially be used as a rapid screening method for AGR2-related disease.
In conclusion, we describe a previously unrecognised autosomal recessive disease which is caused by biallelic variants in the AGR2 gene, likely acting via a LoF mechanism. Paediatric patients presenting a CF-like phenotype should be tested for AGR2. Our findings are relevant for the early genetic diagnosis and timely clinical management of the patients—acting as the first step to unravelling the pathophysiology of this disease.
留言 (0)