
記住我


Synthetic cannabinoid receptor agonists (SCRAs) represent a large portion of detected new psychoactive substances (NPS) globally, accounting for 29% of the 1,047 NPS identified between 2009 and 2019 (1). Commonly sold as herbal blends, such as “Spice,” “K2,” and “Black Mamba” which are consumed by smoking, SCRAs are part of a conscious effort by manufacturers and retailers to mimic the effect of Δ9-tetrahydrocannabinol (THC, 1, Figure 1), the primary intoxicating compound in cannabis (2–6). Several mass intoxication events have been observed over the past decade, with clinically significant impact on patients including psychosis, seizure, respiratory failure, encephalopathy, necrotizing pancreatitis, acute kidney injury, and death (3, 7–31).
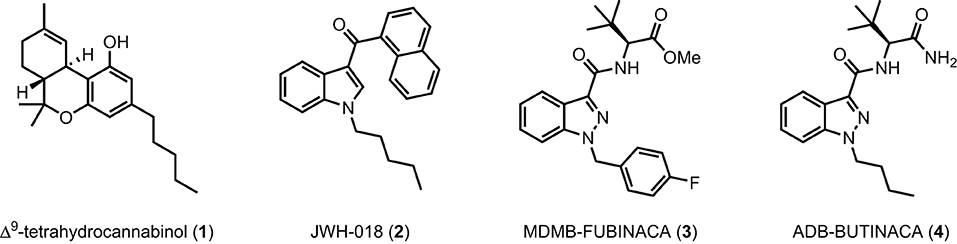
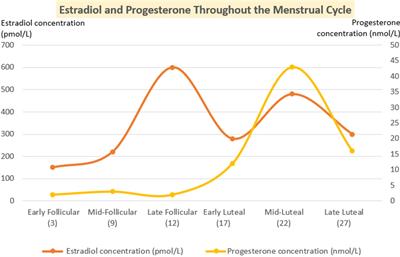
FIGURE 1
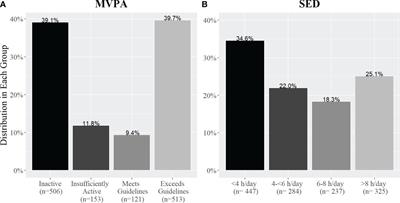
Figure 1. Δ9-THC (1), JWH-018 (2), and selected SCRAs MDMB-FUBINACA (3) and ADB-BUTINACA (4).
SCRAs primarily target the endocannabinoid system, specifically as agonists of centrally expressed cannabinoid 1 receptors (CB1), to provide users with a high analogous to cannabis, although these compounds generally display equal or better affinity and potency at CB1 compared with THC. However, unlike THC, SCRAs such as JWH-018 (2), MDMB-FUBINACA (3), and ADB-BUTINACA (4) generally act as high efficacy agonists at CB1 resulting in significantly greater intoxication of users, and display differing profiles of tolerance, dependence, and withdrawal (32).
Modern SCRAs typically consist of amino acid-derived indole- and indazole-3-carboxamide type scaffolds, similar to compounds disclosed by Pfizer in a series of patents in 2009 (33–38). Appearance of NPS containing modification of these scaffolds has seemingly been a result of incoming legislation bringing detected compounds under national and international control, while attempting to retain CB1 activity. Recent approaches observed include scaffold hopping, fluorination, nitrogen walking, alkyl chain contraction, and homologation, as well as the coupling of different amino acid residues to the substituted heterocyclic cores. These modifications have led to a significant structural evolution of contemporary SCRAs from the late 2000s, with equivalent or increased potency at CB1 (39–41).
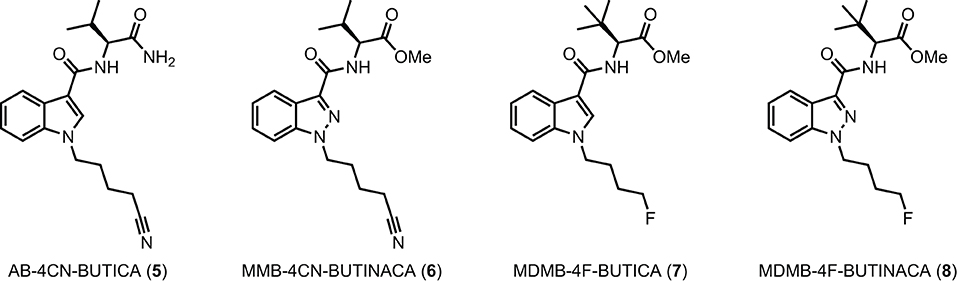
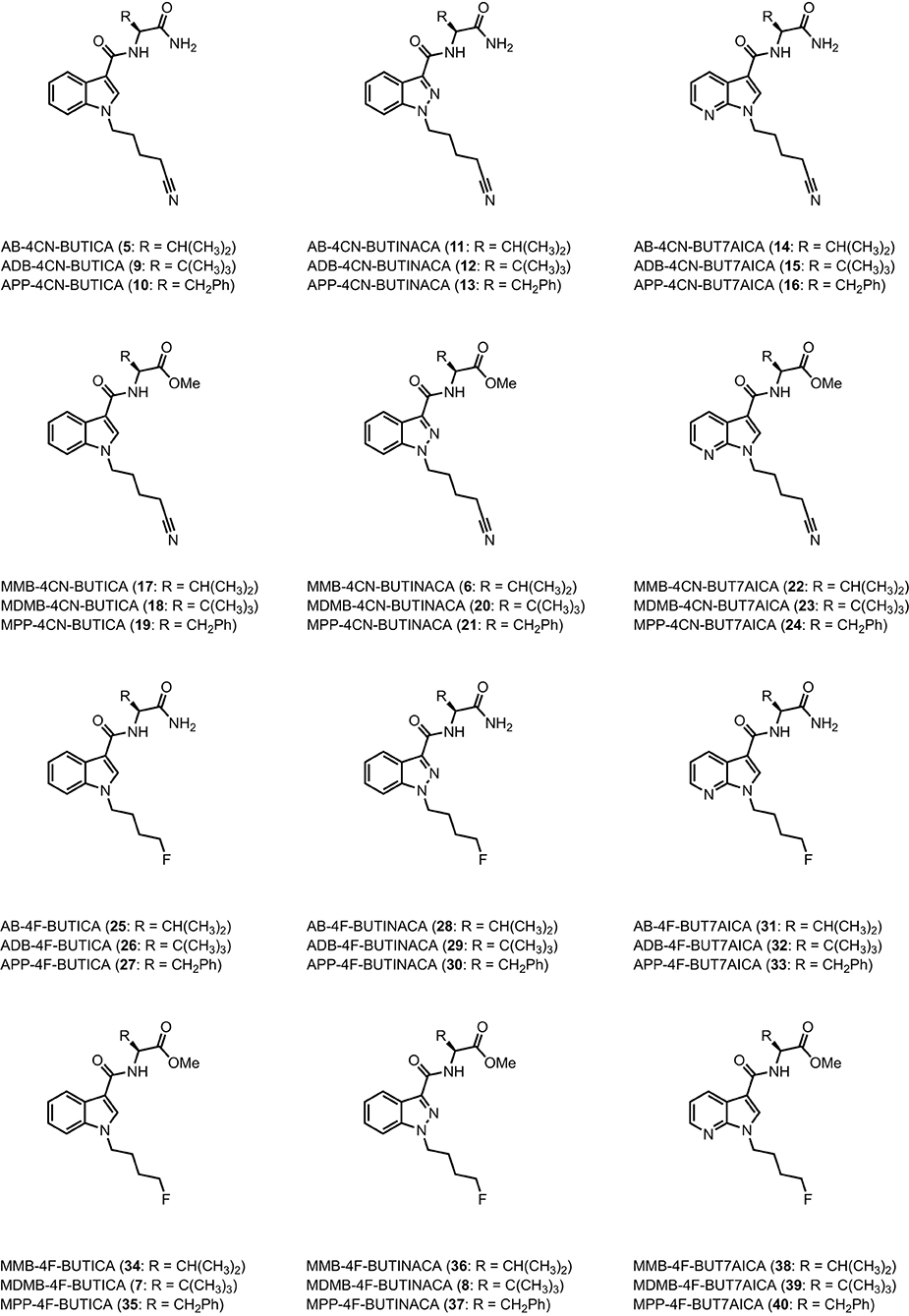
The utility of proactive synthesis programs to detect emerging NPS has recently facilitated the detection of two new SCRAs bearing the 4-cyanobutyl tail moiety. The indole and indazole derivatives AB-4CN-BUTICA (5) and MMB-4CN-BUTINACA (6, a.k.a. AMB-4CN-BUTINACA), respectively, were identified in Alabama, facilitated by proactive reference standard generation and toxicological screening by our laboratories (Figure 2) (42, 43). The 4-fluorobutyl compounds MDMB-4F-BUTICA (7) and MDMB-4F-BUTINACA (8) were also recently detected, with the latter involved in a suicide, as well as multiple fatal intoxications (7, 31, 44–49). With the appearance of these compounds, it is feasible that NPS manufacturers may introduce the 4-fluorobutyl or 4-cyanobutyl subunits to other amino acid-derived indole-, indazole- and 7-azaindole-3-carboxamide SCRAs in future. As such, this paper describes the synthesis, chemical analysis, and pharmacological characterization of the newly detected SCRAs, as well as a series of 32 systematic analogs (5–40, Figure 3). Due to the combinatorial design of the library, direct comparison of the contributions of subunits to the pharmacological profile of the compounds enabled identification of key structure-activity relationships (SARs). In vitro binding and functional data were obtained at both CB1 and CB2, and an in silico docking approach was utilized to explore observed SARs, especially concerning differences in binding between 4-cyanobutyl and 4-fluorobutyl tail moieties. Most compounds displayed high affinity, potency, and efficacy at both CB1 and CB2, suggesting these compounds should be included in NPS monitoring programs. Given the similarity in structure and pharmacological profile of the library evaluated in this work compared to existing SCRAs, these data provide important insights into the potential effect of these compounds in humans, pending evaluation in vivo.
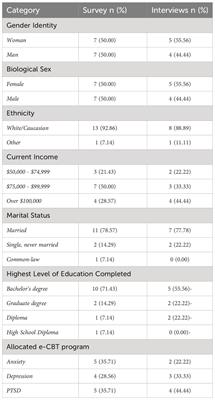
FIGURE 2
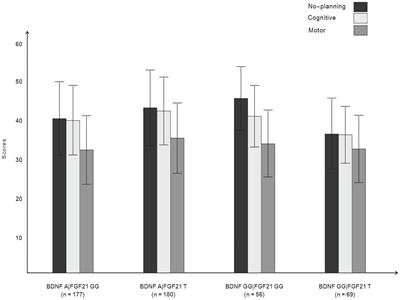
Figure 2. Detected SCRAs AB-4CN-BUTICA (5), MMB-4CN-BUTINACA (6), MDMB-4F-BUTICA (7), and MDMB-4F-BUTINACA (8).
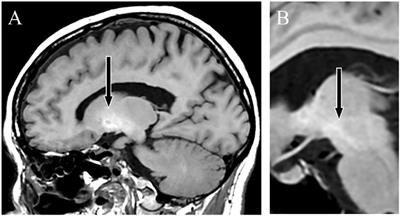
FIGURE 3
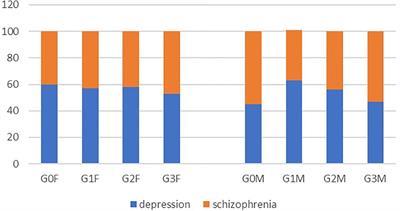
Figure 3. Structures of amino acid-derived 4-cyanobutyl and 4-fluorobutyl-substitued indole, indazole and 7-azaindole SCRAs explored in this study. AB, [(S)-2-amino-3-methylbutanamide]; ADB, [(S)-2-amino-3,3-dimethylbutanamide]; APP, [(S)-2-amino-3-phenylpropanamide]; MMB, [methyl (S)-2-amino-3-methylbutanoate]; MDMB, [methyl (S)-2-amino-3,3-dimethylbutanoate]; MPP, [methyl ((S)-2-amino-3-phenylpropanoate]; 4-CN-BUT, 4-cyanobutyl; 4F-BUT, 4-fluorobutyl; ICA, indole-3-carboxamide; INACA, indazole-3-carboxamide; 7AICA, 7-aza-indole-3-carboxamide.
Experimental General chemical synthesis detailsAll reactions were performed under an atmosphere of nitrogen unless otherwise specified. Methyl 1H-indazole-3-carboxylate (42) and methyl 1H-pyrrolo[2,3-b]pyridine-3-carboxylate (43) were purchased from Fluorochem LTD (Derbyshire, UK) and used as received. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC•HCl) was purchased from Oakwood Chemical (Estill, SC, USA). Deuterated solvents (CD3OD, CDCl3, and DMSO-d6) were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA). Unless otherwise stated, all other reagents and solvents used for this manuscript were obtained from Sigma-Aldrich/Merck (Castle Hill, NSW, Australia) and used as purchased. Analytical thin-layer chromatography was performed using Merck aluminum-backed silica gel 60 F254 (0.2 mm) plates (Merck, Darmstadt, Germany), which were visualized using shortwave (254 nm) UV fluorescence. Flash chromatography was performed using a Biotage Isolera Spektra One and Biotage SNAP KP-Sil silica cartridges (Uppsala, Sweden), with gradient elution terminating at the solvent combination indicated for each compound (vide infra). Melting point ranges (m.p.) were measured in open capillaries using a Stuart SMP50 Automated melting point apparatus (Cole-Palmer, Staffordshire, UK) and are uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded at 298 K using an Agilent 400-MHz spectrometer (Santa Clara, CA, USA), or a Bruker NEO 300 MHz NMR spectrometer (Billerica, MA, USA). The data are reported as chemical shift (δ ppm) relative to the residual protonated solvent resonance, relative integral, multiplicity (s = singlet, brs = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet), coupling constants (J Hz), and assignment. High resolution mass spectrometry (HRMS) data were collected using an Agilent LC 1260-QTOF/MS 6550 (Santa Clara, CA, US) or a Bruker Solarix 2xR 7T Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. A methanolic extract of each pure standard was run using an electrospray ionization (ESI) source in automated MS/MS (information dependent acquisition) mode. Accurate mass for the parent ion and its corresponding mass error expressed in parts per million (ppm) are reported. LC-MS/UV data were acquired using a Shimadzu (Kyoto, Japan) Nexera LC-30AD UHPLC system coupled to a Shimadzu SPD-20AV photo diode array detector and a Shimadzu LCMS-8040 triple-quadrupole mass spectrometer, equipped with an electrospray ionization (ESI) source. Compounds were dissolved in a 90:10 mixture of 0.1% formic acid in water and methanol, and placed in the UHPLC autosampler that was maintained at 8°C. Elution was performed in gradient mode with an Agilent (CA, USA) Zorbax XDB-C18 column (2.1 × 50 mm, 3.5 μm particle size), held at 50°C, with a 10 μL injection volume. The mobile phases were 0.1% formic acid in water (A) and methanol (B). Mobile phase composition was held at 10% B until 0.5 min, then steadily increasing to 100% B at 2.5 min, holding until 3 min, and then re-equilibrating at 10% B for a total run time of 4 min. UV absorbance was measured from 190 to 800 nm. UV data from blank injections were subtracted to account for mobile phase absorbance and background noise. Single stage mass spectra (MS1) were collected in positive ESI mode using a single quadrupole (Q3) scanning from m/z 100–600. All data were processed using Shimadzu LabSolutions (v 5.89) software. Fourier-transform infrared spectroscopy (FTIR) spectra were collected using a Bruker Alpha II ATR FTIR spectrophotometer. Precursors 44–51, and compounds 6, 7, 11, 17, and 22 were synthesized and used as is, as previously described (50, 51). Synthetic procedures and characterization data (1H and 13C NMR, melting point, Rf, FTIR, HRMS, UV, and LCMS) can be found in the supporting information.
In vitro binding evaluationHuman embryonic kidney (HEK) cells expressing either human CB1 receptors N-terminally tagged with pplss (preprolactin signal sequence) and 3HA (3x haemagglutinin) epitopes or human CB2 receptor N-terminally tagged with 3HA were harvested in 5 mM EDTA in PBS, and “P2” membranes were prepared in sucrose buffer as previously described (52, 53). Protein content was estimated using a BioRad (Hercules, CA) DC protein assay (modified Lowry assay). For binding assays, radioligand ([3H]-CP55,940, PerkinElmer, Waltham, MA, USA), non-radiolabeled drugs, and P2 membrane preparations were diluted in binding buffer (50 mM HEPES pH 7.4, 1 mM MgCl2, 1 mM CaCl2, 2 mg/mL NZ-origin BSA, MP Biomedicals, Santa Ana, CA, USA) and dispensed into 96-well, polypropylene V-well plates (Hangzhou Gene Era Biotech Co Ltd, Zhejiang, China) in a final reaction volume of 200 μL (membranes were dispensed last). Final radioligand concentration was 1 nM for all assays. Protein content was 3 μg/point for pplss-3HA-hCB1 HEK membranes, and 2 μg/point for 3HA-hCB2 HEK membranes for the assays using type A harvest plate (PerkinElmer, GF/C filters, 1.2 μm pores), these plates were discontinued during the course of this study, and replaced with type B harvest plates (Merk Millipore, GF/C filters, 1.2 μm pores). As these plates appear to retain a smaller proportion of the protein, membrane content was increased to 8 μg/point for CB1 membranes, and 3 μg/point for CB2 membranes. When all components had been dispensed, the V well plate was sealed and incubated for 1 h at 30°C. During the incubation, the 96 well harvest plate was treated with 0.1% w/v branched polyethyleneimine (PEI; Sigma Aldrich) in H2O. Immediately prior to washing, PEI was washed through the filters using a vacuum manifold (Pall Corporation, Port Washington, NY) and all wells were washed once with ice cold wash buffer (50 mM HEPES pH 7.4, 500 mM NaCl, 1 mg/mL BSA). Equilibrated binding mixture was then transferred to the harvest plate under vacuum, and samples washed through. For the assays using type A harvest plate, binding wells were rinsed once with wash buffer and transferred to the harvest plate, and then wells were washed three more times with 200 μL of wash buffer. The plate was then removed, and filters allowed to dry overnight. The next day, the plate bottom was sealed, and 50 μL of Ultima Gold XR scintillation fluid (PerkinElmer) was dispensed to each well and the plate loaded into the 96 well “rigid” cassette. For the assays using type B harvest plate, samples were rapidly washed 4 times with 100 μL of wash buffer. The bottom of the plate was then removed, and filters allowed to dry overnight. The next day, the plate was inserted into a 96 well “rigid” cassette. The base of the cassette was sealed and 50 μL of scintillation fluid was added to each well. The top of the plates were sealed prior to loading into a Wallac MicroBeta2® TriLux Liquid Scintillation Counter (PerkinElmer). Scintillation was detected after a 30 min delay, for 2 min per well. Counts were corrected for detector efficiency. Data were then exported and analyzed in GraphPad Prism v9 (GraphPad Software Inc., La Jolla, CA, USA). Ki was determined through fit of “Competition binding- One site fit Ki” in GraphPad prism using a Kd of 3.50 nM for the radioligand for the assays using type A harvest plate; while for the assays using harvest plate B, Kd was 3.58 nM for binding at CB1 and 1.162 nM for binding at CB2 (Kd was determined empirically using homologous competition assay under matching conditions). Log Ki were determined for at least three independent experiments (maximum 6) and combined to determine mean pKi ± SEM reported in Tables 1, 2. Assays for compounds 5, 6, 10–12, 14, 15, 17, 18, 20–22, 24, 29, 34 used type A harvest plate. Assays for compounds 7–9, 13, 16, 19, 23, 25–28, 30–33, 35–40 used type B harvest plates.
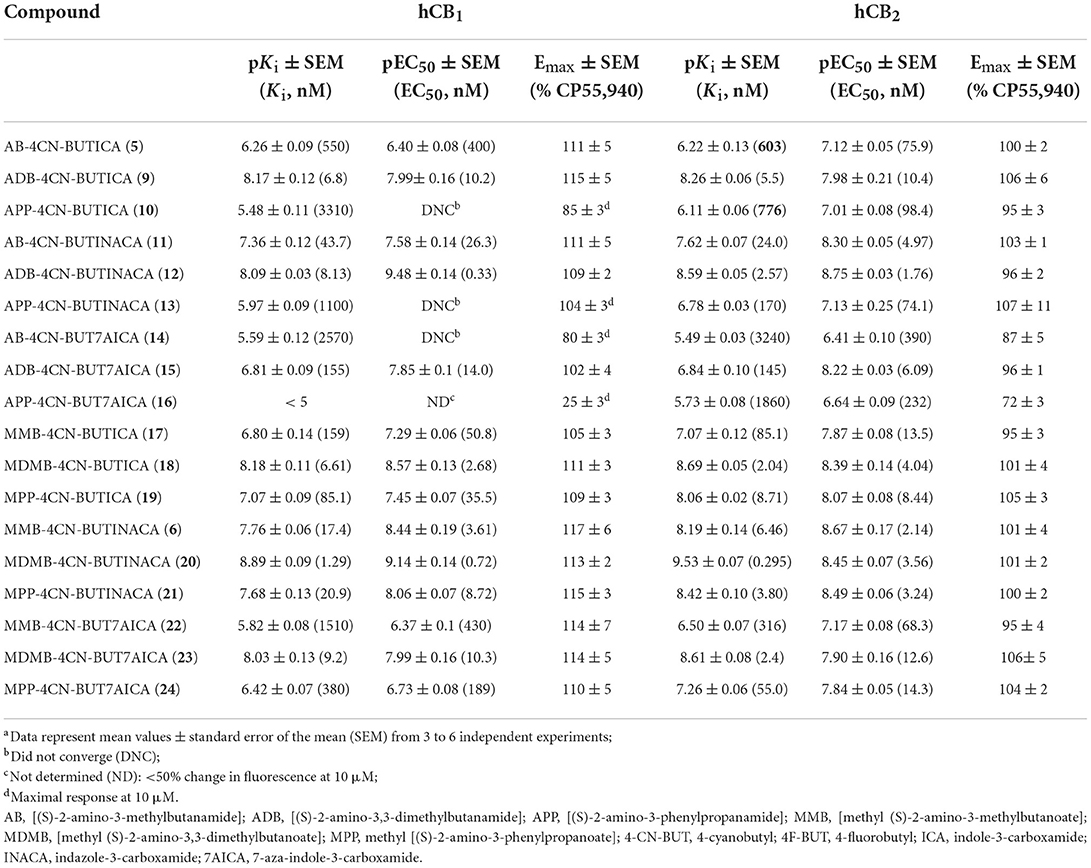
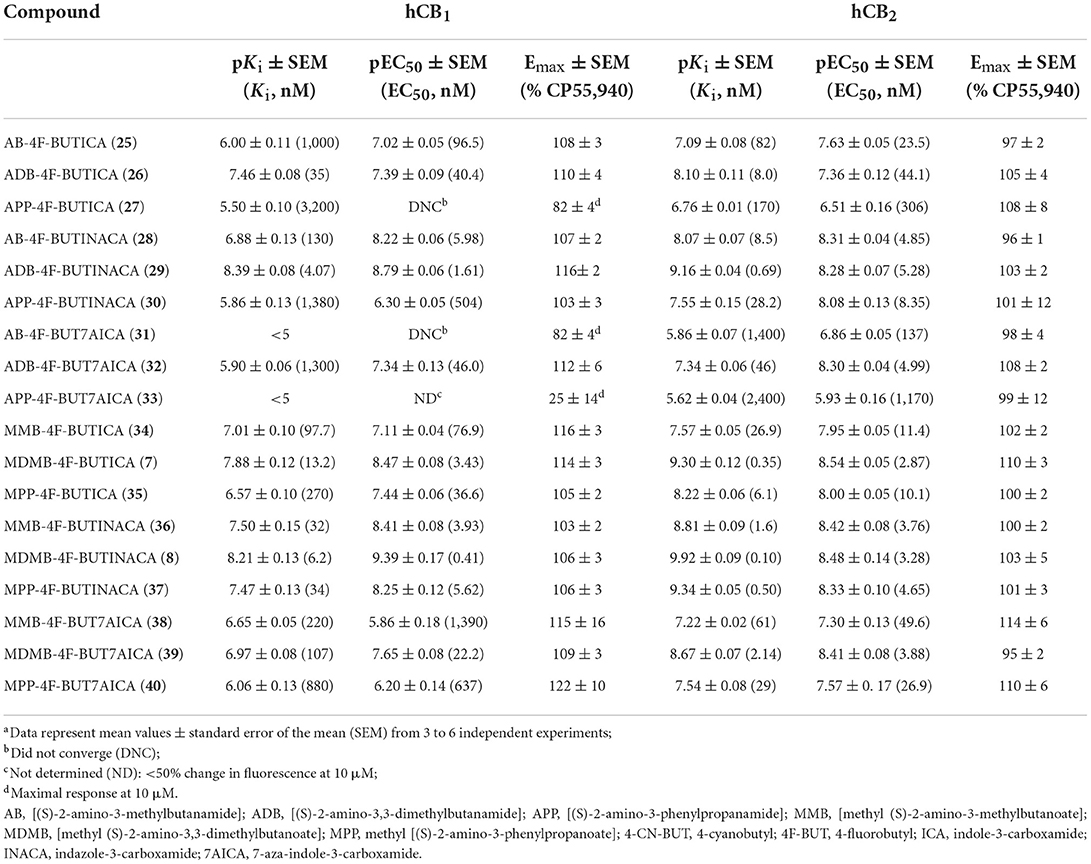
TABLE 1
Table 1. Affinities and functional activities of 4-cyanobutyl derived SCRAs at hCB1 and hCB2.
TABLE 2
Table 2. Affinities and functional activities of 4-fluorobutyl derived SCRAs at hCB1 and hCB2.
In vitro functional evaluationMouse AtT20 FlpIn adenocarcinoma cells stably transfected with human CB1 or CB2 were cultured in DMEM containing 10% FBS, 100 U penicillin/streptomycin, and 80 μg/mL of hygromycin, as previously described (54). Cells were passaged at 80% confluency, cells for assays were grown in 75 cm2 flasks and used at 90% confluence. The day before the assay cells were detached from the flask with trypsin/EDTA (Sigma Aldrich) and resuspended in 10 mL of Leibovitz's L-15 media (L-15) supplemented with 1% FBS, 100 U penicillin/streptomycin, and 15 mM glucose. The cells were plated in volume of 90 μL in black walled, clear bottomed 96-well microplates (Corning, Corning, NY, USA) and incubated overnight at 37°C in ambient CO2. Membrane potential was measured using a Membrane Potential Assay Kit (blue) from Molecular Devices (San Jose, CA, USA), as described previously (55, 56). The dye was reconstituted with assay buffer [145 mM NaCl, 22 mM HEPES, 0.338 mM Na2HPO4, 4.17 mM NaHCO3, 0.441 mM KH2PO4, 0.407 mM MgSO4, 0.493 mM MgCl2, 1.26 mM CaCl2, 5.56 mM glucose (pH 7.4)]. Prior to the assay, cells were loaded with 90 μL/well of the dye solution without removal of the L-15. Plates were then incubated at 37°C in ambient CO2 for 60 min. Fluorescence was measured using a FlexStation 3 (Molecular Devices) microplate reader with cells excited at a wavelength of 530 nm and emission measured at 565 nm. Baseline readings were taken every 2 s for at least 2 min, at which time either drug or vehicle was added in a volume of 20 μL. The background fluorescence of cells without dye or dye without cells was negligible. Changes in fluorescence were expressed as a percentage of baseline fluorescence after subtraction of the changes produced by vehicle (DMSO, final concentration was no more than 0.1%) addition. Concentration-response data were analyzed using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA), using a four-parameter non-linear regression to fit concentration-response curves on data from 3 to 6 independent experiments.
Molecular modeling Protein preparationThe cryo-EM structures of the CB1 receptor (PDB: 6N4B) and the CB2 receptor (PDB: 6KPF) were retrieved from RCSB PDB (57–59). The structures were prepared with Maestro's Protein Preparation Wizard as follows (60). The G proteins and cholesterol were removed, leaving only the CB1 and CB2 receptor and their cognate ligands. The preparation process consisted of assigning bond orders, adding hydrogens, generation of disulfide bonds, generation of missing side chains using Prime, generating het states using Epik at pH 7.4 ± 1.0, and deleting water molecules beyond 3 Å from het groups (61, 62). The hydrogen bonding network was optimized, the pKa values of the protein were predicted using PROPKA, and target pH value was set at 7.4 (63). Lastly, the protein structure was minimized using the OPLS4 force field where RMSD of the atom displacement for terminating the minimization was set as 0.3 Å (64).
Ligand preparationLigands were prepared using LigPrep to generate energy minimized 3D structures (65). OPLS4 force field was used for minimization. Epik was used to generate all possible ionized states at pH 7.4 ± 1.0.
Ligand dockingA receptor grid was generated using Glide, with a van der Waals radius scaling factor of 1.0 and a partial charge cutoff at 0.25 (66). The binding site was defined by the centroid of the cognate ligand for each structure. The Van der Waals scaling factor for the ligands was set to 0.80 with a partial charge cut-off at 0.15. The precision was set to Extra Precision (XP) with flexible ligand sampling. Nitrogen inversions and ring conformations were sampled and Epik state penalties were added to the docking scores. The ligand cores were restricted to the position of the cognate ligand by their maximum common substructure and post docking minimization was performed. Poses that had the lowest RMSD between the 4-fluorobutyl and 4-cyanobutyl analogs were taken for direct comparison.
Strain calculationsThe internal strain of ligands in their bound and unbound states was calculated using MacroModel (65). The 4RDDD solvation forcefield was used, a constant 4.00 kcal/mol energy offset selected, a penalty scale factor of 0.25 and Cartesian restraints with a bound state half-width of 0.3 Å and a 120 kcal/mol/Å2 force constant.
Binding site mappingThe binding site regions for both the CB1 and CB2 structures were evaluated using SiteMap (67). SiteMap was run in evaluate mode using the structures cognate ligand and a 5 Å buffer. A minimum of 15 site points per binding site was set, using the “more restrictive” definition of hydrophobicity and a standard grid. Site maps were cropped at 4 Å from the nearest site point.
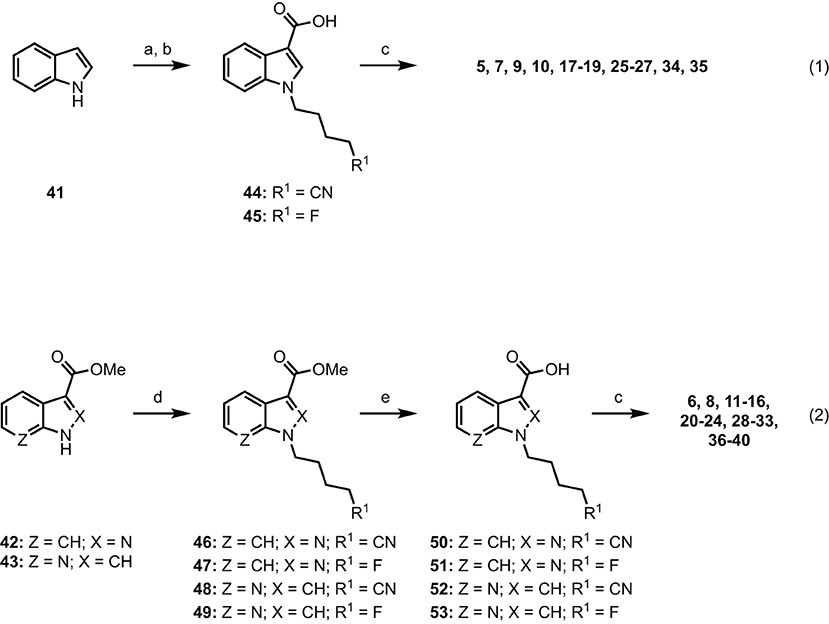
Results and discussion SynthesisIndole-based SCRAs were derived from indole (41), with a convenient one-pot alkylation and trifluoracetylation procedure employed to give intermediate N-alkyl-5-(3-(2,2,2-trifluoroacetyl)-1H-indoles before subsequent hydrolysis gave N-alkyl-1H-indole-3-carboxylic acids 44 and 45 in good yield (Scheme 1, eq. 1). This was then subjected to 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/1-hydroxybenzotriazole (HOBt)-mediated amide coupling with the appropriate amino acid derivatives to furnish the final indole derived compounds (5, 7, 9, 10, 17–19, 25–27, 34, 35). The indazole and 7-azaindole derivatives were synthesized using a similar sequence from methyl indazole- or methyl-7-azaindole-3 carboxylates 42 and 43 (Scheme 1, eq. 2). Thus, alkylation with the appropriate alkylbromide and sodium hydride provided methyl N-alkyl-indazole- or methyl N-alkyl-7-azaindole-3 carboxylates 46–49 before hydrolysis to give the corresponding carboxylic acids 50–53. In the same manner as described previously, EDC/HOBt-mediated amide coupling furnished N-alkyl-indazole- or -alkyl-7-azaindole-3-carboxylamides 6, 8, 11–16, 20–24, 28–33, 36–40 (6, 50, 51, 54, 68, 69). Synthetic procedures and characterization data (1H and 13C NMR, melting point, Rf, FTIR, HRMS, and LCMS) for all novel compounds can be found in the supporting information.
SCHEME 1
Scheme 1. Reagents and conditions: (a) (i) NaH, alkylbromide, DMF, 0 °C–rt, 1 h; (ii) (CF3CO)2O, 0 °C–rt, 2 h; (b) KOH, MeOH, PhMe, reflux, 2 h; (c) EDC•HCl, HOBt•H2O, amino acid•HCl, Et3N, DMF, rt, 18 h, 29–99%. (d) NaH, alkylbromide, DMF, 0°C–rt, 24 h, 81%; (e) 1 M aq. NaOH, MeOH, rt, 48 h, 98%.
In vitro binding affinity trends for 5–40Following our previous efforts in the characterization of existing SCRAs, in vitro binding data were obtained via a competitive radio binding assay in HEK293 cells stably transfected with human cannabinoid receptor 1 (hCB1) or human cannabinoid receptor 2 (hCB2) (6, 50, 54, 68, 69). Ligand affinity (pKi) was determined based on extent to which compounds displaced the tritiated standard [3H]CP55,940 (Tables 1, 2). Except for terminal amide bearing 7-azaindoles SCRAs 38, 33 and 16, all compounds displayed micromolar to sub-nanomolar affinity for CB1 receptors (pKi = 8.89 ± 0.09–5.48 ± 0.11). Similar or increased CB2 affinity was observed in almost all cases (pKi = 9.92 ± 0.09–5.49 ± 0.03), with several examples exhibiting sub-nanomolar affinities for this receptor subtype (20, pKi = 9.53 ± 0.07; 29, pKi = 9.16 ± 0.04; 7, pKi = 9.30 ± 0.12; 8, 9.92 ± 0.09; 37, pKi = 9.34 ± 0.05), which represent the highest affinities in the series. Across the 4-cyanobutyl and 4-fluorobutyl tail substituents, no clear trend was apparent for CB1 except within methyl tert-leucinate (18, 20, 23 > 7, 8, 39) and methyl phenylalaninate (35, 37, 40 > 19, 21, 24) subgroups, whereby 4-cyanobutyl and 4-fluorobutyl substitution, respectively, conveyed increased affinity, albeit slightly. Only 4-cyanobutyl bearing 15 (CB1 pKi = 6.81 ± 0.09) showed affinity an order of magnitude greater than its 4-fluorobutyl analog 32 (CB1 pKi = 5.90 ± 0.06). Conversely, 4-fluorobutyl derivatives were in general equal or better ligands at CB2. Variation of the headgroup within the indazole series (AB: 28 > 11; ADB: 29 > 12; APP: 30 > 13; MMB: 36 > 4; MDMB: 8 ≈20; MMP: 37 ≈21) highlights this trend. A clear structure-affinity trend within the core scaffold was observed, with indazoles (CB1, pKi = 8.89 ± 0.09–5.48 ± 0.11; CB2, pKi = 9.53 ± 0.07–6.78 ± 0.03) providing the best affinity to both CB1 and CB2, followed closely by indoles (CB1, pKi = 8.18 ± 0.11–5.50 ± 0.10; CB2, pKi = 9.30 ± 0.12–6.22 ± 0.13). Finally, 7-azaindoles (CB1, pKi = 8.03 ± 0.13– < 5; CB2, pKi = 8.67 ± 0.07–5.49 ± 0.03) exhibited reduced affinity compared with the other groups, often by an order of magnitude. This is in keeping with previous studies detailing related indole, indazole and 7-azaindole carboxamides (36, 51, 54). Notably, the indazole core was required to achieve sub-nanomolar affinity for CB2 (i.e., 20, 29, 8, 37) except in the case of 7, whereby high affinity was maintained with combination of an indole core, and importantly, the methyl tert-leucinate head group. In each core and tail sub-group, the tert-leucine derivatives (MDMB > ADB) were consistently the best ligands for both CB1 and CB2, followed by phenylalanine methyl esters (MPP), which were better than both valine derivatives (MMB > AB), and finally, phenylalaninamides (APP). For example, within the indole set, a tert-leucinate conferred the highest CB1 affinity (18, pKi = 8.18 ± 0.11), which was reduced by an order of magnitude for the corresponding phenylalaninate (19, pKi = 7.07 ± 0.09), and further halved for valine derivatives (17, pKi = 6.80 ± 0.14). An alternate order was observed for the corresponding terminal amides of tert-leucine (9, pKi = 8.17 ± 0.12), valine (5, pKi = 6.26 ± 0.09) and phenylalanine (10, pKi = 5.48 ± 0.11), which conferred one of the lowest affinities in the set. Affinities for CB2 followed the same general trend, although phenylalaninamides exhibited affinities up to an order of magnitude greater compared to CB1 (e.g., 13, CB1 pKi = 5.48 ± 0.11; CB2 pKi = 6.78 ± 0.03). Indeed, APP derivatives were not well-tolerated at CB1 and thus modest selectivity for CB2 (4- to 50-fold) was observed in this class (i.e., 10, 33, 16, 27, 30).
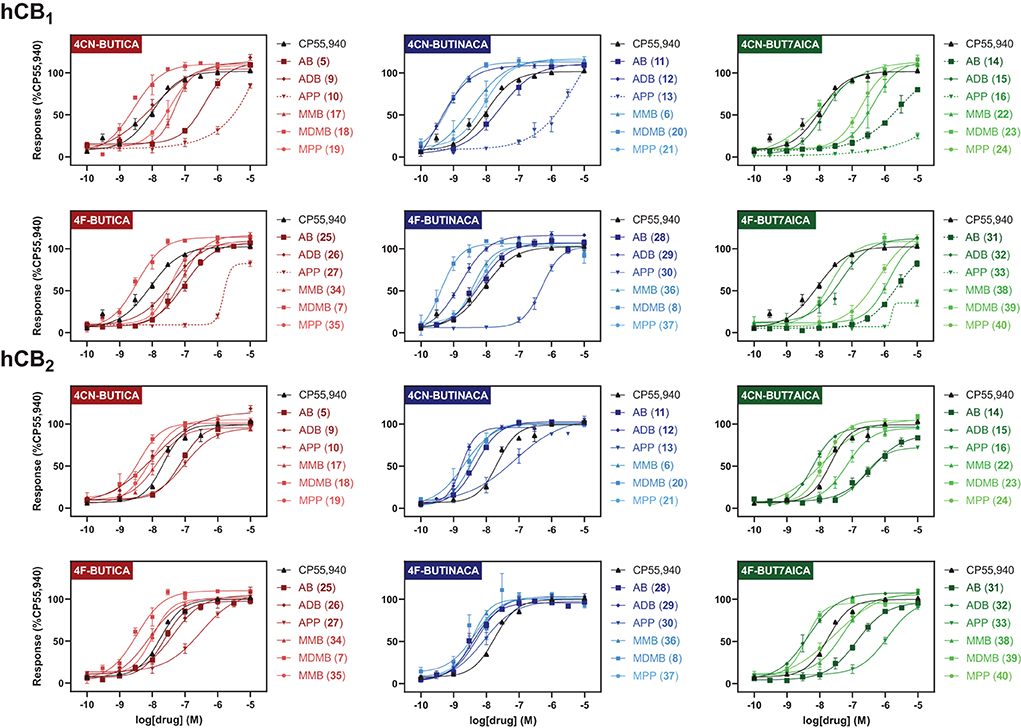
In vitro CB1 and CB2 functional characterization of 5–40SARs for compounds 5–40 were also investigated via a fluorescence-based functional assay using AtT20 cells transfected with hCB1 and hCB2. This method measures change in membrane potential resulting from Gβγ-coupled activation of inwardly rectifying potassium channels (GIRKs). The full concentration-response curves of 5–40 are shown in Figure 4, with potencies and efficacies referenced to 1 μM CP55,940 in Tables 1, 2. Most compounds, except for selected phenylalaninamide and/or 7-azaindole derivatives (10, 13, 16, 27, 33, and 38), had maximal efficacy (Emax = 102–122%), and exhibited nanomolar to sub-nanomolar potencies (pEC50 = 9.48 ± 0.14–6.30 ± 0.05) at CB1. In general, this group activated CB2 with equal or increased potency (pEC50 = 8.67 ± 0.17–6.41 ± 0.10) and efficacy, albeit, with no ligands active at sub-nanomolar levels. As with other SCRAs, these compounds are generally similar or greater potency, but more efficacious than THC (pEC50 = 6.76 ± 0.09 and Emax = 58% ± 3% at CB1, and 32% ± 1% at 30 μM at CB2) (36). Potency compared with THC is also similar (pKi = 8.09 ± 0.02 and 7.50 ± 0.07 at CB1 and CB2, respectively) (70).
FIGURE 4
Figure 4. Functional activities of compounds 5–40 at hCB1 and hCB2, relative to CP55,940. Efficacies are normalised to maximal response of 1 μM CP55,940. Vertical sets indicate the same heterocyclic core (left = indole; centre = indazole; right = 7-azaindole). Graphs are grouped by tail, with rows indicating alkyl tails, either 4-cyanobutyl (rows 1 and 3), or 4-fluorobutyl (rows 2 and 4). Dashed lines indicate where curve could not be fit. Data is expressed as means ± SEM from 3 to 6 independent experiments.
Effect of 4-cyanobutyl and 4-fluorobutyl substituents on CB1 and CB2 activationOf the 3 chemical modifications investigated in the present study, variation of the tail substituent had the least significant impact on potency and efficacy. The 4-cyanobutyl and 4-fluorobutyl derivatives commonly displayed similar potency at both CB1 and CB2, for example in the tert-leucinate subgroup (18, 20, 23 ≈7, 8, 39, respectively), or conversely, exhibited differing potency with no discernable preference for either tail group. Indeed, both tail substituents produced active ligands with sub-nanomolar potency for CB1 (20: pEC50 = 9.14 ± 0.14; 8: pEC50 = 9.39 ± 0.17) and low nanomolar potency for CB2 (20: pEC50 = 8.45 ± 0.07; 8: pEC50 = 8.48 ± 0.14). These data are consistent with the corresponding structure-affinity trends; however, binding is not predictive of relative potency in some cases. For example, MDMB-4F-BUTINACA (8) exhibits activation (pEC50 = 9.39 ± 0.17) an order of magnitude greater than its affinity (pKi = 8.21 ± 0.13) for CB1, whilst for CB2, the opposite is true (pEC50 = 8.48 ± 0.14; pKi = 9.92 ± 0.09).
Effect of indole, indazole and 7-azaindole core on CB1 and CB2 activationIn the present study, the nature of the heterocyclic core greatly impacts potency, giving rise to the same trends as previously documented (51, 54, 71). Rank order potency of activity at CB1 closely mirrored the observed binding affinities, whereby indazoles (pEC50 = 9.39 ± 0.17–6.31 ± 0.05) demonstrated equal or greater potency to the corresponding indoles (pEC50 = 8.57 ± 0.13– < 5), which were, again, more potent than the corresponding 7-azaindoles (pEC50 = 8.05 ± 0.21– < 5). In all cases, the indazole core was required to produce sub-nanomolar potency (pEC50 = 9.39 ± 0.17, 9.48 ± 0.14, 9.14 ± 0.14) observed for compounds 8, 12, and 20, respectively. While the same general trend is realized for activation of CB2 (indazoles: pEC50 = 8.75 ± 0.03–7.00 ± 0.36; indoles: pEC50 = 8.54 ± 0.05–6.49 ± 0.2; 7-azaindoles: pEC50 = 8.41 ± 0.08–5.92 ± 0.16), differentiation of heterocyclic cores is less pronounced for this receptor, with the MPP subgroup (21 > 19 > 24) highlighting this effect. Notably, this is not apparent for MDMB derivatives (7 ≈8 ≈39), whereby all core motifs conferred similar potency.
Effect of amino acid derived head groups on CB1 and CB2 activationAs described previously, tert-leucine derived compounds (MDMB/ADB) are commonly more potent than the corresponding valine (ADB/AB) species at CB1, despite differing only by a single methyl group (36, 51, 54, 69, 71, 72). In line with this is the greater potencies of MDMB/ADB vs. MMB/AB analogs for indole (18, 9 > 5, 17), indazole (20, 12 > 6, 11) and 7-azaindole (23, 15 > 22, 14) sub-groups observed here. Further, MDMB- and ADB-4F-BUTINACA (8 and 29) represent two of only three substrates with sub-nanomolar potency, and indeed the highest potencies observed for CB1. While the methyl esters of phenylalanine (MPP) were commonly equipotent or greater than valine (AB and MMB) analogs, unlike with tert-leucine derivatives, switching to the terminal amide (APP) was detrimental to potency at CB1. Specifically, when combined with an indole (10 and 27) or 7-azaindole core (16 and 33), the APP substituent produced submaximal agonist activity at the highest concentration tested (10 μM), whereas full efficacy was maintained with the indazole core (i.e., 13: Emax = 104%). In terms of CB2, structure-activity trends align closely with CB1; however, with all phenylalaninamides (APP) except APP-4CN-BUT7AICA (16: Emax = 73%, pEC50 = 6.63 ± 0.11) having full agonist effect with moderate potency. As with binding, these findings indicate a CB2 receptor subtype selectivity for the phenylalaninamide group. In general, the SARs described here corroborate the observed structure-affinity trends for the compounds in the present study, as well as those previously found (54).
In silico molecular docking studyTo rationalize the novel SARs observed in vitro at CB1 and CB2, we performed docking for each of the screened ligands using the cryo-EM structures of the CB1 (PDB ID: 6N4B) and the CB2 (PDB ID: 6PT0) receptors retrieved from the Research Collaboratory for Structural Bioinformatics Protein Databank (RCSB PDB) (57–59). These agonist-bound structures were chosen for the similarity between our compounds and the cognate ligands MDMB-FUBINACA (6N4B) and WIN 55,212-2 (6PT0).
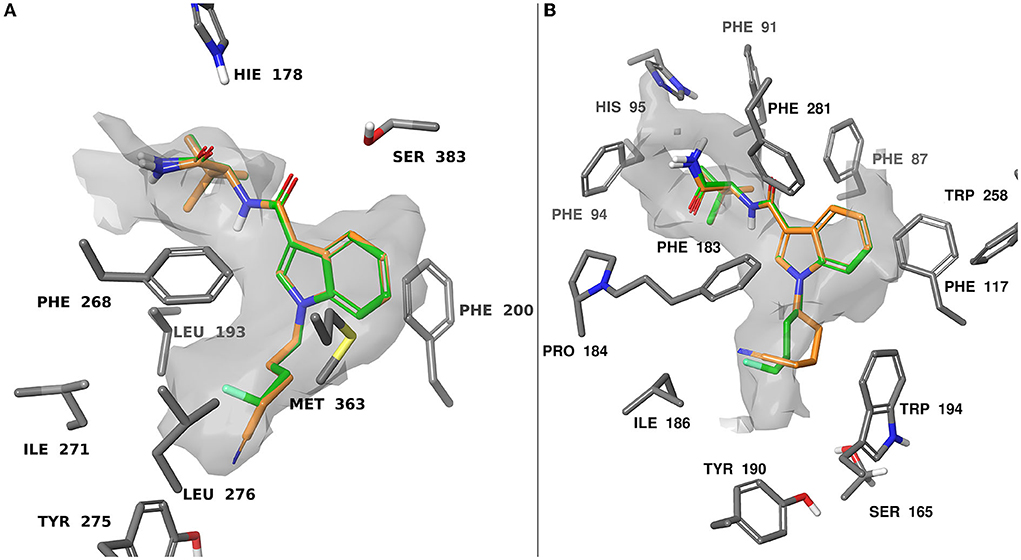
When docking at CB1 there were π-π interactions between each of the ligands and residues PHE200 and PHE268. The flexible 4-fluorobutyl and 4-cyanobutyl tail groups occupied a hydrophobic pocket comprised of MET363, LEU276, TYR275, LEU193, and ILE271. There is also a common polar interaction between the amide linker carbonyl and SER383, as well as the terminal carbonyl and HIE178.
Docking at CB2 was dictated by hydrophobic interactions between the core and PHE117, and PHE183 residues. The flexible tail occupied a pocket comprised of TRP194, ILE186, TYR190, and SER165. The amide and ester head groups occupied a pocket comprised of PHE91, PHE94, HIS95, and PRO184. Ligands bearing a terminal amide had the potential for H-bonding with the backbone carbonyl of PHE183. Pairs of docking poses for ligands that differed by tail substitution were selected for strain rescoring (see SI, figures S105–116 for all calculated docking poses). This allowed us to investigate the SAR described in the in vitro section, wherein many 4-fluorobutyl ligands showed higher affinity for the CB2 receptor than their 4-cyanobutyl counterparts. Given the lack of formal intermolecular interactions possible for this substituent, the effect is likely due to the size-constraints of the binding pocket for CB2 as compared with CB1. Figure 5 shows an overlay of compounds 9 and 26 at both CB1 and CB2, as well as the computed hydrophobic regions of the binding site. Relative to CB1, the CB2 binding site region occupied by the 4-fluorobutyl or 4-cyanobutyl tail is significantly constrained. This was observed by Sitemap calculation on the active site of CB1 and CB2 receptors as illustrated in Figure 5. This may force the longer and more rigid 4-cyanobutyl tail into a higher strain conformation to adopt the predicted binding pose. The strain energy of the ligands in their bound state relative to their unbound state was calculated, and the values compared for analogous ligands (see SI Table T1). The mean difference and standard error between the docked 4-cyanobutyl and 4-fluorobutyl ligands were 0.220 ± 0.400 kcal/mol at CB1 and 2.552 ± 0.468 kcal/mol at CB2.
FIGURE 5
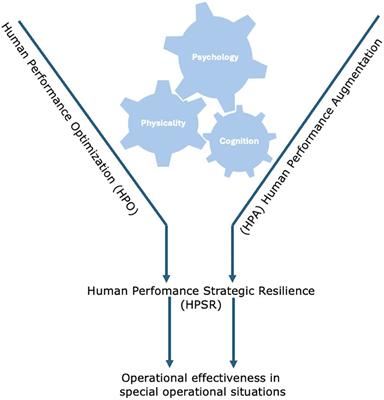
Figure 5. Computationally predicted binding modes of 9 (orange) and 26 (green) overlayed at CB1(A), PDB:6N4B and CB2(B), PDB:6PT0 (57, 58). The hydrophobic regions of the binding site (grey) were evaluated with SiteMap (67). Glide docking was performed using the Schrodinger computational chemistry suite (66).
ConclusionsThe detected SCRAs AB-4CN-BUTICA, MMB-4CN-BUTINACA, MDMB-4F-BUTICA, MDMB-4F-BUTINACA, as well as a series of 32 analogs, were synthesized, characterized, and evaluated in vitro using a radioligand binding assay and a functional membrane potential assay at both human CB1 and CB2. These data confirm that AB-4CN-BUTICA, MMB-4CN-BUTINACA, MDMB-4F-BUTICA, MDMB-4F-BUTINACA are potent and efficacious cannabinoid receptor ligands. Most analogs in the present study, barring the APP (phenylalaninamide) derivatives, were also potent and efficacious cannabinoid ligands. SAR trends observed here were consistent with those previously described for with respect to head group and core group contributions to ligand activity (30, 36, 50, 54, 69, 72). The increased affinity for 4-fluorobutyl derivatives at CB2 is likely to arise from increased strain of the 4-cyanobutyl tail in the hydrophobic tail pocket. Given these pharmacological data, availability of precursory chemical building blocks, ease of synthesis, and structural similarity to previous SCRAs, the compounds evaluated in this study should be monitored as potential emerging NPS in the marketplace.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributionsES and JL synthesized and characterized compounds under the supervision of AA and SB. In vitro binding data were obtained by SC under the supervision of MG. In vitro functional characterization was performed by RB and CF under the supervision of EC and by TF and HZ under the supervision of MS and MC. In silico docking experiments were performed by JM under the supervision of FL and DH. HRMS data were obtained by RE under the supervision of RG. ES, JM, IM, EC, MC, MG, AA, and SB conceived the experiments. ES, JM, EC, AA, and SB wrote the manuscript. All authors reviewed drafts of the manuscript and approved the final version.
FundingParts of this work were supported NHMRC Project Grant 1161571 and Brain and Mind Centre Research Development Grant awarded to SB. ES, RB, JM, CF, IM, EC, AA, and SB are supported by the Lambert Initiative for Cannabinoid Therapeutics, a philanthropic research program based at the University of Sydney. ES, JM, and TF are supported by an Australian Government Research Training Scholarships through the University of Sydney and Macquarie University respectively.
AcknowledgmentsThis research was facilitated by access to Sydney Mass Spectrometry, a core research facility at the University of Sydney, with the authors acknowledging Chianna Dane for obtaining several HRMS spectra. The authors would like to thank Rebecca Gordon and Dr. Richard Kevin from the Lambert Initiative for Cannabinoid Therapeutics for obtaining LCMS and UV spectra. The authors from the University of Sydney would like to acknowledge the Gadigal people of the Eora Nation as the traditional custodians of the land on which they work, and where this research was conducted. These authors would like to pay their respects those who have cared and continue to care for Country, to elders past, present, and emerging.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.1010501/full#supplementary-material
References2. Halter S, Mogler L, Auwarter V. Quantification of herbal mixtures containing Cumyl-PEGACLONE-is inhomogeneity still an issue? J Anal Toxicol. (2020) 44:81–5. doi: 10.1093/jat/bkz028
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Brandehoff N, Adams A, McDaniel K, Banister SD, Gerona R, Monte AA. Synthetic cannabinoid “Black Mamba” infidelity in patients presenting for emergency stabilization in Colorado: a P SCAN Cohort. Clin Toxicol. (2018) 56:193–8. doi: 10.1080/15563650.2017.1357826
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gatch MB, Forster MJ. Delta9-Tetrahydrocannabinol-like effects of novel synthetic cannabinoids found on the gray market. Behav Pharmacol. (2015) 26:460–8. doi: 10.1097/FBP.0000000000000150
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Banister SD, Arnold JC, Connor M, Glass M, McGregor IS. Dark classics in chemical neuroscience: delta(9)-tetrahydrocannabinol. ACS Chem Neurosci. (2019) 10:2160–75. doi: 10.1021/acschemneuro.8b00651
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Van Rafelghem B, Covaci A, Anseeuw K, van Nuijs ALN, Neels H, Mahieu B, et al. Suicide by vaping the synthetic cannabinoid 4F-MDMB-BINACA: cannabinoid receptors and fluoride at the crossroads of toxicity? Forensic Sci Med Pathol. (2021) 17:684–8. doi: 10.1007/s12024-021-00424-7
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Manini AF, Krotulski AJ, Schimmel J, Allen L, Hurd YL, Richardson LD, et al. Respiratory failure in confirmed synthetic cannabinoid overdose. Clin Toxicol. (2021) 60:524–26. doi: 10.1080/15563650.2021.1975734
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Hermanns-Clausen M, Muller D, Kithinji J, Angerer V, Franz F, Eyer F, et al. Acute side effects after consumption of the new synthetic cannabinoids AB-CHMINACA and MDMB-CHMICA. Clin Toxicol. (2018) 56:404–11. doi: 10.1080/15563650.2017.1393082
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Kusano M, Zaitsu K, Taki K, Hisatsune K, Nakajima J, Moriyasu T, et al. Fatal intoxication by 5F-ADB and diphenidine: detection, quantification, and investigation of their main metabolic pathways in humans by LC/MS/MS and LC/Q-TOFMS. Drug Test Anal. (2018) 10:284–93. doi: 10.1002/dta.2215
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Giorgetti A, Busardo FP, Tittarelli R, Auwarter V, Giorgetti R. Post-mortem toxicology: a systematic review of death cases involving synthetic cannabinoid receptor agonists. Front Psychiatry. (2020) 11:464. doi: 10.3389/fpsyt.2020.00464
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Lam RPK, Tang MHY, Leung SC, Chong YK, Tsui MSH, Mak TWL. Supraventricular tachycardia and acute confusion following ingestion of e-cigarette fluid containing AB-FUBINACA and ADB-FUBINACA: a case report with quantitative analysis of serum drug concentrations. Clin Toxicol. (2017) 55:662–7. doi: 10.1080/15563650.2017.1307385
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Papadopoulos EA, Cummings KR, Marraffa JM, Aldous KM, Li L, Ahmad N. Reports of adverse health effects related to synthetic cannabinoid use in New York State. Am J Addict. (2017) 26:772–5. doi: 10.1111/ajad.12636
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Schep LJ, Slaughter RJ, Hudson S, Place R, Watts M. Delayed seizure-like activity following analytically confirmed use of previously unreported synthetic cannabinoid analogues. Hum Exp Toxicol. (2015) 34:557–60. doi: 10.1177/0960327114550886
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Schwartz MD, Trecki J, Edison LA, Steck AR, Arnold JK, Gerona RR. A common source outbreak of severe delirium associated with exposure to the novel synthetic cannabinoid ADB-PINACA. J Emerg Med. (2015) 48:573–80. doi: 10.1016/j.jemermed.2014.12.038
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Thornton SL, Akpunonu P, Glauner K, Hoehn KS, Gerona R. Unintentional pediatric exposure to a synthetic cannabinoid (AB-PINACA) resulting in coma and intubation. Ann Emerg Med. (2015) 66:343–4. doi: 10.1016/j.annemergmed.2015.05.021
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Tyndall JA, Gerona R, De Portu G, Trecki J, Elie MC, Lucas J, et al. An outbreak of acute delirium from exposure to the synthetic cannabinoid AB-CHMINACA. Clin Toxicol. (2015) 53:950–6. doi: 10.3109/15563650.2015.1100306
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Barcelo B, Pichini S, Lopez-Corominas V, Gomila I, Yates C, Busardo FP, et al. Acute intoxication caused by synthetic cannabinoids 5F-ADB and MMB-2201: A case series. Forensic Sci Int. (2017) 273:e10–14. doi: 10.1016/j.forsciint.2017.01.020
PubMed Abstract | CrossRef Full Text | Google Scholar
25. McQuade D, Hudson S, Dargan PI, Wood DM. First European case of convulsions related to analytically confirmed use of the synthetic cannabinoid receptor agonist AM-2201. Eur J Clin Pharmacol. (2013) 69:373–6. doi: 10.1007/s00228-012-1379-2
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Buser GL, Gerona RR, Horowitz BZ, Vian KP, Troxell ML, Hendrickson RG, et al. Acute kidney injury associated with smoking synthetic cannabinoid. Clin Toxicol. (2014) 52:664–73. doi: 10.3109/15563650.2014.932365
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R. “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N Engl J Med. (2017) 376:235–42. doi: 10.1056/NEJMoa1610300
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Springer YP, Gerona R, Scheunemann E, Shafer SL, Lin T, Banister SD, et al. Increase in adverse reactions associated with use of synthetic cannabinoids - anchorage, Alaska, 2015-2016. MMWR Morb Mortal Wkly Rep. (2016) 65:1108–11. doi: 10.15585/mmwr.mm6540a4
留言 (0)