
記住我
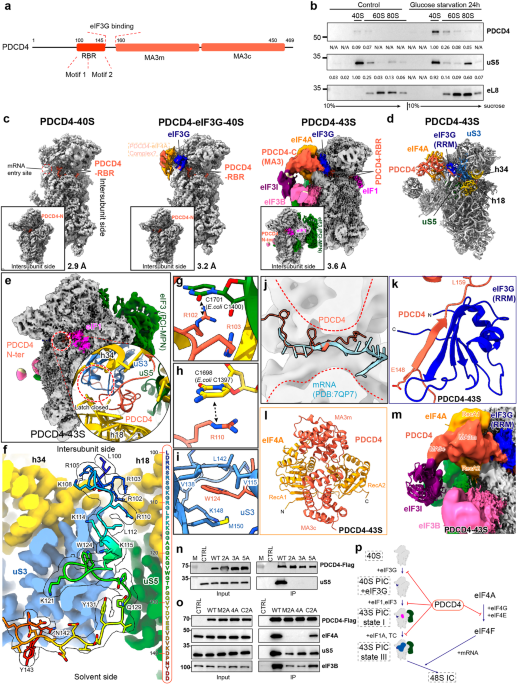

While widespread mitochondrial outer membrane permeabilization (MOMP) generally seals the fate of stressed cancer cells as it elicits caspase-dependent mechanisms that regulate the kinetics and immunological manifestations of cell death, partial MOMP driving sublethal caspase activation has been associated with cancer cell survival in the context of accrued genomic instability. Recent findings delineate a novel caspase-independent mechanism linking sublethal MOMP to the acquisition of a persister phenotype coupled to chemoresistance and accrued metastatic potential.
Most therapeutic regimens currently employed for the clinical management of cancer, including conventional chemotherapeutics, radiation therapy and targeted anticancer agents exert cytotoxic effects by activating intrinsic apoptosis.1 In this setting, unrepairable organellar damage elicits signal transduction cascades that culminate with widespread mitochondrial outer membrane permeabilization (MOMP) downstream of BCL2 associated X, apoptosis regulator (BAX), BCL2 antagonist/killer 1 (BAK1) and/or BCL2 family apoptosis regulator BOK oligomerization at the outer mitochondrial membrane, a process that is tonically inhibited by antiapoptotic members of the same protein family, such as BCL2, BCL2 like 1 (BCL2L1, best known as BCL-XL) and MCL1, BCL2 family member.2
Besides annihilating bioenergetic and metabolic mitochondrial functions, widespread MOMP results in the cytosolic accumulation of mitochondrial components including cytochrome c, somatic (CYCS), which is a potent activator of apoptotic caspases like caspase 3 (CASP3), ultimately sealing the cellular fate. Conversely, MOMP affecting only a minority of mitochondria has been shown to promote tumor progression downstream of a CASP3-dependent pathway culminating with accrued genomic instability as a consequence of DNA damage by DNA fragmentation factor subunit beta (DFFB, best known as CAD).3 Recent data delineates a CASP3-independent mechanism whereby sublethal MOMP coupled to partial cytosolic CYCS promotes chemoresistance and accrued metastatic potential upon the activation of the integrated stress response (ISR) via eukaryotic translation initiation factor 2 alpha kinase 1 (EIF2AK1, best known as HRI) and activating transcription factor 4 (ATF4).4 These findings have long-reaching implications as they identify a novel mechanism through which sublethal apoptotic signaling may favor the survival of drug-tolerant, persistent cancer cells ultimately driving disease relapse and treatment failure (Fig. 1).
Fig. 1: Sublethal apoptotic signaling drives chemoresistance and metastatic spread independent of caspases.
Partial MOMP resulting in sublethal activation of apoptotic caspases such as caspase 3 (CASP3) has been previously linked to accrued genomic instability and accelerated tumor progression via CASP3-dependent mechanisms. Recent findings demonstrate that sublethal MOMP also ignites a CASP3-independent pathway of adaptation to stress that involves CYCS, EIF2AK1 (best known as HRI) and ATF4. This mechanism endows cancer cells surviving sublethal apoptotic signaling with a drug-tolerant, persistent phenotype associated with accrued metastatic spread. Whether chronic, indolent innate immune signaling elicited by partial MOMP contributes to this process by favoring the establishment of the metastatic niche remains to be elucidated.
By harnessing two distinct inhibitors of antiapoptotic BCL2 proteins, namely ABT-737 and S63845, at sublethal doses, Kalkavan and colleagues successfully established a transient persister state (PS) in human lung adenocarcinoma and colorectal cancer cells. Such a PS was characterized by increased resistance to BCL2 inhibition and chemotherapy in vitro (but an increased sensitivity to activators of ferroptosis, a regulated variant of necrosis),1 by genetic signatures of the epithelial-to-mesenchymal transition (EMT) and NF-κB signaling, by a minor decrease in proliferation rate, as well as by accrued metastatic potential in vivo (upon intravenous or orthotopic inoculation into immunodeficient mice). Importantly, co-deletion of BAX, BAK1 and BOK not only restored the sensitivity of human lung adenocarcinoma cells driven into the PS to BCL2 inhibitors and their resistance to ferroptosis inducers, but also limited their metastatic behavior, demonstrating the mechanistic dependence of the PS on proapoptotic BCL2 family members.4
Single-cell RNA sequencing confirmed that the PS as elicited by suboptimal BCL2 inhibition was associated with transient genetic signatures of the EMT and NF-κB signaling, as it pointed to the activation of the ISR, a cell-wide, integrated response to adverse conditions centered around the endoplasmic reticulum (ER) and mitochondria.5,6 Accordingly, BCL2 inhibitors caused the HRI-dependent phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1, best known as eIF2α), causing ATF4 stabilization coupled to a generalized shift from cap-dependent to internal ribosome entry site (IRES)-mediated protein translation. Importantly, this pathway could be prevented by BAX, BAK1 and BOK co-deletion, but not by the knockout of genes encoding key activators of CASP3 downstream of MOMP, such as apoptotic peptidase activating factor 1 (APAF1) and CASP9.4 These data point to the existence of a caspase-independent mechanism linking sublethal MOMP to the ISR via HRI and ATF4.
Importantly, CYCS turned out to physically bind, hence activating, HRI upon release from mitochondria exposed to BCL2 inhibitors at sublethal doses. In line with this notion, CYCS deletion as well as the knockout of holocytochrome c synthase (HCCS), which is involved in heme loading onto immature CYCS, prevented ATF4 activation and the acquisition of a PS in lung cancer cells undergoing partial MOMP. Moreover, EIF2AK1 or ATF4 deletion similarly reduced the resistance of lung cancer cells to BCL2 inhibition as they limited their metastatic potential upon intravenous inoculation into immunocompromised mice. At least in part, ATF4 appeared to promote chemoresistance and the PS by driving BAX downregulation, potentially (but not necessarily) linked to decreased translation of BAX (which does not contain an experimentally validated IRES)7 in the context of eIF2α phosphorylation.4 Corroborating the pathophysiological relevance of these findings, HRI levels were found to be higher in multiple solid tumors as compared to adjacent healthy tissues, and to negatively correlate with overall survival in a cohort of patients with lung cancer.4
Altogether, these data define a novel, caspase-independent mechanism through which cancer cells exposed to sublethal challenge may acquire phenotypic and behavioral traits that are detrimental for the patient, including chemoresistance and pronounced metastatic potential (Fig. 1). As MOMP is also associated with the release of mitochondrial components that elicit inflammatory responses, such as mitochondrial DNA (mtDNA),8,9 it will be interesting to dissect the immunological component of these findings in immunocompetent tumor models. On the one hand, potent MOMP-driven inflammatory responses may indeed limit metastatic dissemination in vivo as a consequence of accrued immunosurveillance.8 On the other hand, chronic, indolent inflammatory responses may instead favor metastasis downstream of immunoevasion and formation of optimal metastatic niches.8 In this context, it is worth noting that Kalkavan and collaborators detected signatures of NF-κB signaling in persister cells,4 and that (1) NF-κB is activated by cyclic GMP-AMP synthase (cGAS) upon recognition of cytosolic mtDNA,8 (2) the prototypical NF-κB target gene interleukin 6 (IL6) has a functional IRES,7 and (3) IL6 has previously been implicated in accrued metastatic dissemination to some organs.10 Whether IL6 and/or other cytokines produced downstream of MOMP are at play in the accrued metastatic potential elicited by BCL2 inhibitors at sublethal doses remains to be experimentally verified. Irrespectively, the data by Kalkavan and colleagues nicely delineate yet another scenario whereby sublethal stress renders cancer cells resilient to therapy and increasingly aggressive. These data have far-reaching implications for the development of efficacious anticancer regimens based on BCL2 inhibitors and other MOMP inducers.
留言 (0)