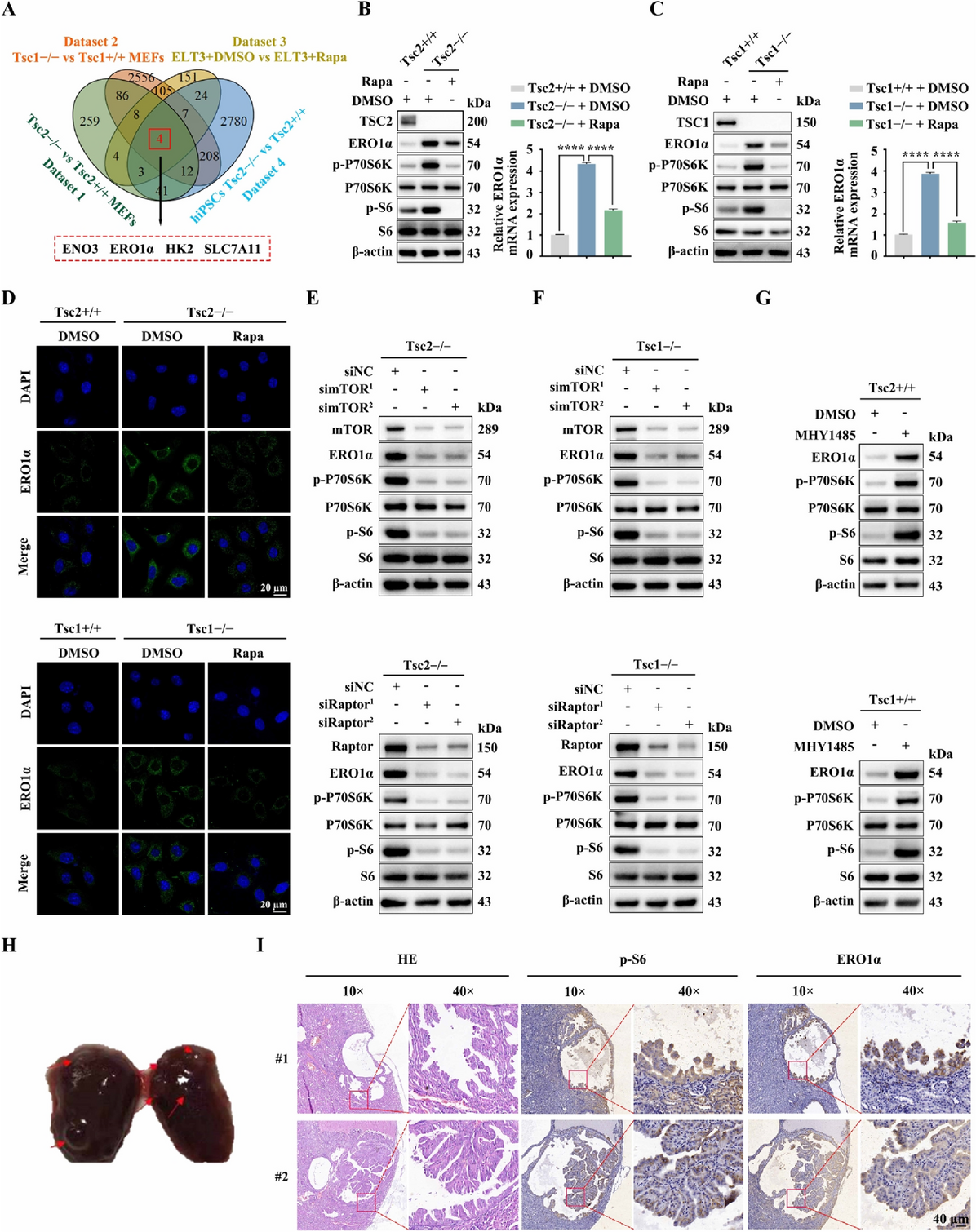
Patient samples
Human adrenocortical tissue samples were collected under the clinical protocol entitled “Prospective comprehensive molecular analysis of endocrine neoplasms” (Clinical Trial Registration number NCT01005654). The ethical approval was granted by the Institutional Review Board, National Cancer Institute, NIH, and the NIH Office of Human Subject Research. All participants provided written informed consent.
qHTS and combination matrix screening
The National Center for Advancing Translational Sciences (NCATS) Pharmaceutical Collection (NPC) and the Mechanism Interrogation PlatE (MIPE) library, which in total consist of 4,991 small-molecule drugs and investigational compounds, were screened against SW13 and NCI-H295R cell lines. Cell viability was measured using a luciferase-coupled ATP quantitation assay (CellTiter-Glo®, Promega, Madison, WI). We described the detailed methods of quantitative high-throughput screening in the supplementary section.
A combination matrix screen was performed with a subset of active hits identified by qHTS. Plating of compounds in matrix format using acoustic droplet ejection and numerical characterization of synergy, additivity, and/or antagonism was conducted as described previously [11, 12]. The detailed method of combination matrix screening was described in the supplementary section.
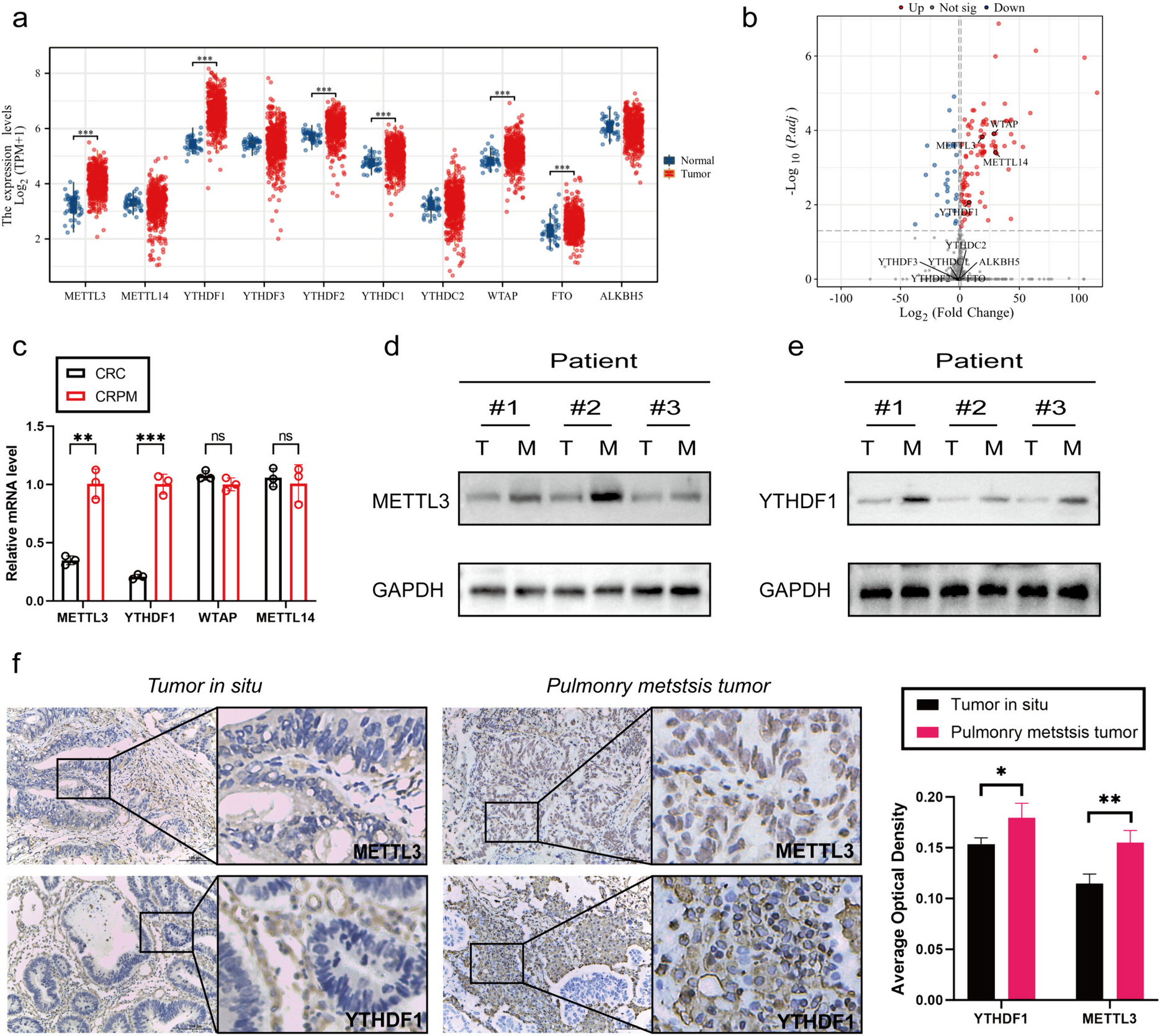
Gene expression profiling
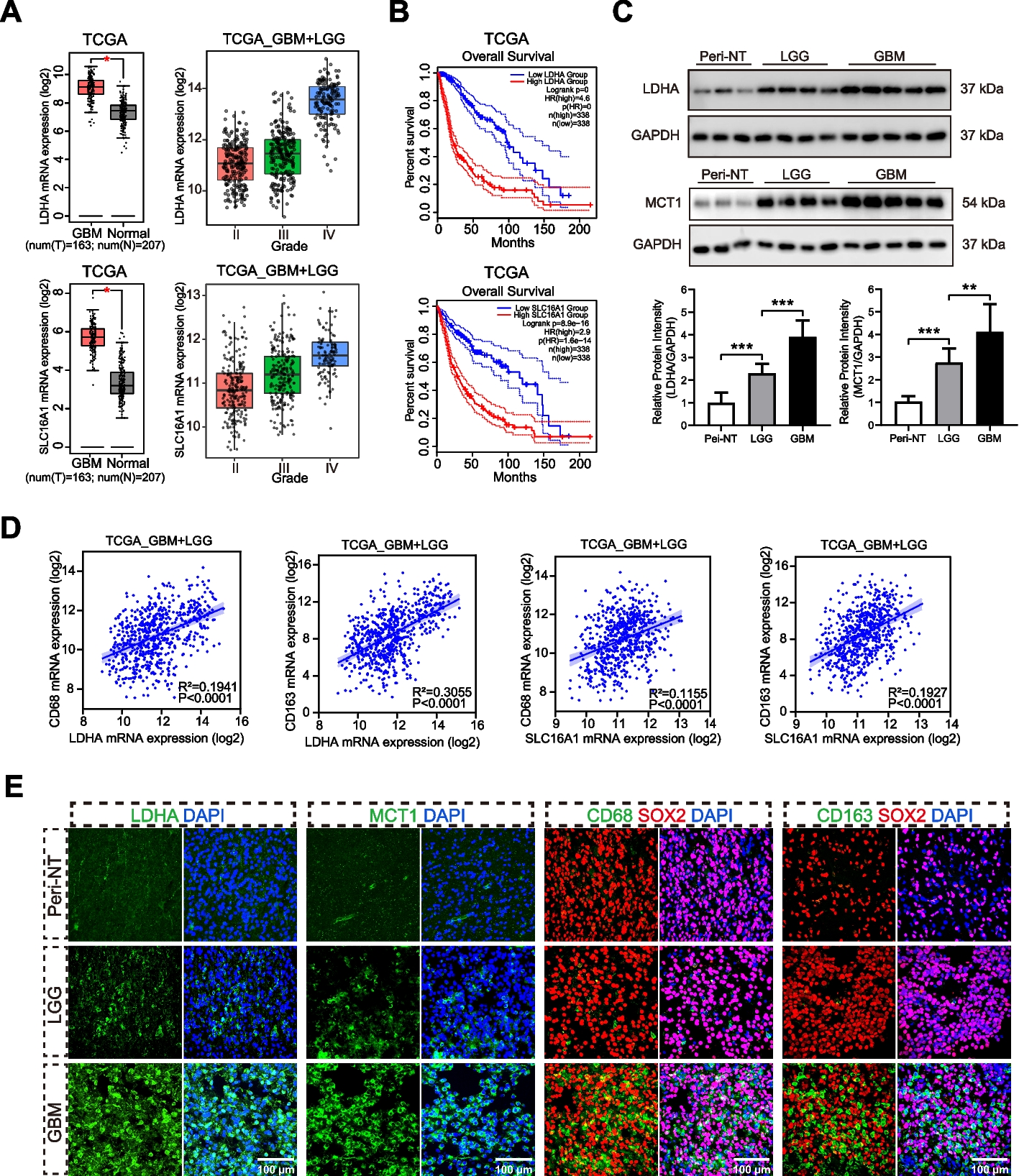
We analyzed publicly available genome-wide expression data from the Gene Expression Omnibus (GEO) (NCBI gene expression and hybridization array data repository) in three cohorts (GSE33371, GSE12368, and GSE90713) to study the differential messenger RNA (mRNA) expression of genes of interest in human ACC samples compared to adrenocortical adenoma (ACA) and normal adrenal cortex (NC) [13]. Using data from TCGA and the European Bioinformatics Institute (E-TABM-311), we analyzed clinicopathologic correlations with mRNA expression of these genes to assess clinical relevance.
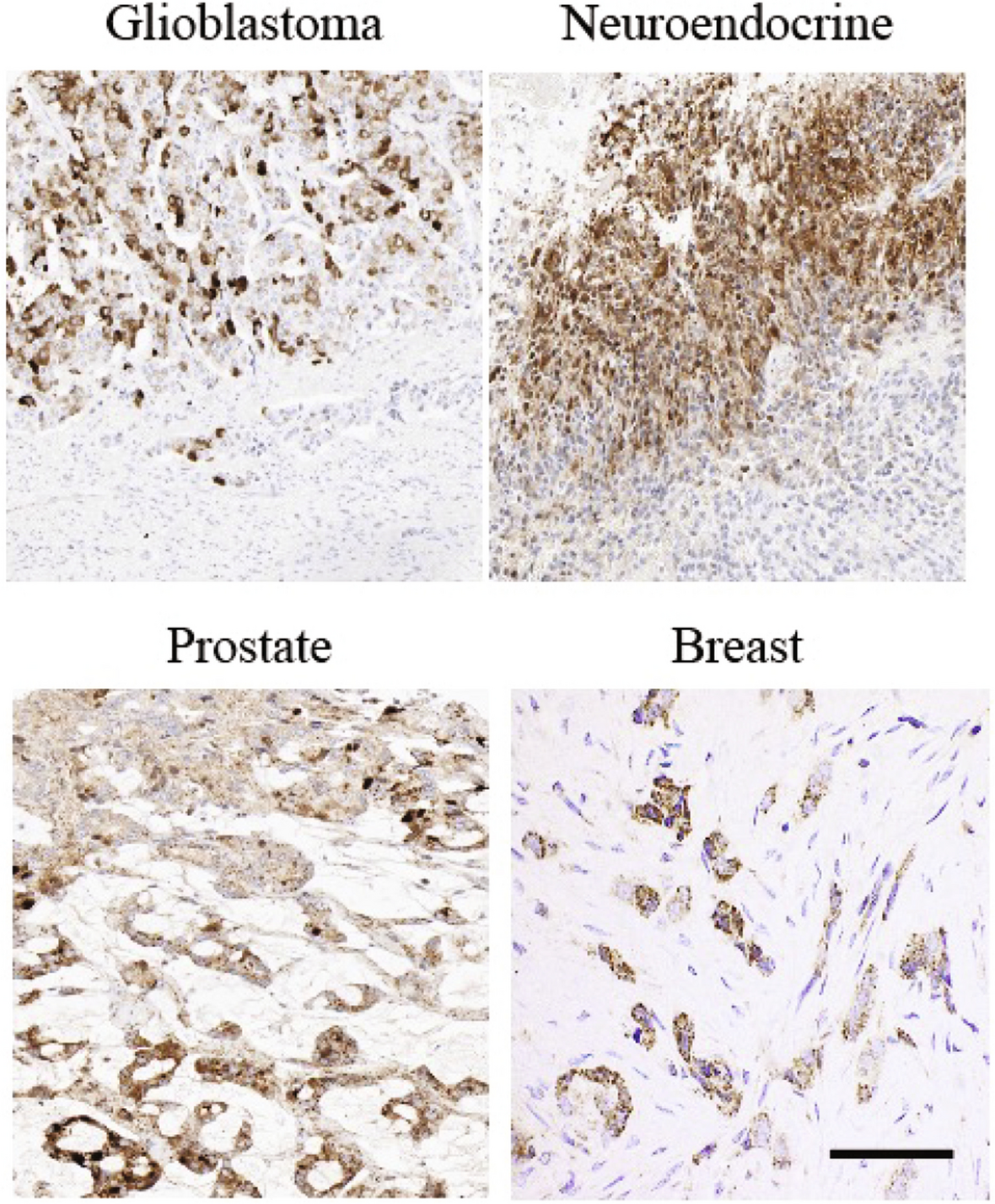
Immunohistochemistry analysis
ACC, ACA patient tissue, and human ACC xenograft tissues were formalin-fixed, embedded in paraffin, and used for immunohistochemistry (IHC) analysis. 5-µm-thick sections were used for hematoxylin and eosin (H&E) and IHC staining according to a previously published protocol [10]. We described the IHC techniques in the supplementary materials section.
Cell lines and culture conditions
Two human ACC cell lines, SW13 and NCI-H295R, were purchased from the American Type Culture Collection™ (Cat # CCL-105, CRL-2128; Manassas, VA, USA) and cultured in 5% CO2 atmosphere at 37 °C in Dulbecco's Modified Eagle Medium (Cat # 11195–065, Thermo Fisher Scientific, MA, USA) supplemented with 2.5% Nu-Serum (Cat # 355100, Corning, MA, USA) and 0.1% Insulin-Transferrin-Selenium (Cat # 41400045, Thermo Fisher Scientific, MA, USA). Cell lines were authenticated by short tandem repeat profiling. We routinely subcultured every 3–5 days, depending on the degree of cell confluence.
NCI-H295R cells used to generate human ACC xenograft were transfected with a linearized pGL4.51[luc2/CMV/Neo] vector (9PIE132, Promega) encoding the luciferase reporter gene luc2 (Photinus pyralis) and maintained in the above medium with up to 500 μg/mL of G-418 antibiotic (11811–023, Gibco, MA, USA) for selection.
Cellular proliferation assay
The effects of drug treatments on cell proliferation were quantitated by CyQuant assay (Cat # C7026, Invitrogen, MA, USA). SW13 (3 × 103) and NCI-H295R (6 × 103) cells were plated in 96-well black plates (Cat # 353219, Costar®, Corning, NY, USA). After 24 h, the culture medium containing vehicle control (dimethyl sulfoxide up to 0.125%), OTS167, RGB-286638, or the combination of OTS167 and RGB-286638 were added at various concentrations. The medium with the drugs or vehicle was replaced every 48 h. Fluorescence intensity was determined using a microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 485 nm/538 nm. We repeated the experiment with consistent results at least three times.
We used the automated computerized algorithm (Chou–Talalay method) to assess the synergistic efficacy. Efficacy indicated by the combination index (CI) was compared to cells treated with a single drug. CI < 1 indicated synergy; CI = 1 indicated an additive effect; and CI > 1, indicated an antagonistic effect [14].
Three-dimensional multicellular aggregates (MCA)
Compared to monolayer cell culture that lacks the tumor microenvironment, MCAs recapitulate the in vivo environment by growing solid, 3-dimensional tumors in vitro more accurately as MCAs contain different areas affected by various degrees of oxygenation, nutrients, and drug exposure [15]. SW13 (6 × 104 cells/0.5 ml) and NCI-H295R (1 × 105 cells/0.5 ml) cells, which form multicellular aggregates (MCA) or tumor spheroids, were plated in ultra-low cluster 24-well plates (Cat No # 3473, Costar®, Corning, NY, USA). The anticancer activity of OTS167, RGB-286638, and the combination of OTS167 and RGB-286638 were tested in MCAs that mimic solid tumors in vitro, in ACC cell lines according to standard protocol. The detailed methods are described in the supplementary materials section.
Clonogenic assay
Cells were seeded in triplicate in 6-well plates (1000 cells/well) and allowed to grow for 7–10 days. The cells were then treated with drug(s) alone or in combination or with the vehicle in complete media for 12 to 14 days. Growth media with vehicle or drug(s) were replaced every 48–72 h. The cells were fixed with 0.4% buffered paraformaldehyde and then stained with 0.5% crystal violet in methanol for 10 min. The colonies were counted and photographed using a ChemiDoc system (Bio-Rad).
Caspase-3/-7 activation assay
To check caspase-3/7-mediated apoptosis, cells were plated in 96-well plates and treated for 24 to 48 h with various concentrations of the drug combination. The caspase-3/-7 activity was measured using the Caspase-Glo® 3/7 assay (Cat # G8091, Promega, USA), according to the manufacturer's instructions. The method of treatment for caspase-3 and -7 activation and analysis are described in the supplementary materials section.
Cell cycle assay
SW13 (3 × 104) and NCI-H295R (2 × 105) cells were plated in a 100-mm dish with 10 mL of culture medium and treated 24 h later. After 24 to72 hours, cells were trypsinized, washed with phosphate-buffered saline (PBS), and fixed with ice-cold 70% ethanol. Cells were then resuspended in PBS with ribonuclease A (100 mg/mL) and propidium iodide (PI) (0.05 mg/mL) for fluorescence-activated cell sorting analysis using the Canto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Data were generated for at least 20,000 events per sample. The cell cycle of the gated PI distribution was analyzed using ModFit software (Verity Software House, Inc., Topsham, ME, USA).
Western blot analysis
Cell lysates were prepared from the cells after treatment with drug(s) or vehicle control. The protein concentration was determined using the Pierce™ BCA assay kit solution (Cat # 23227, Thermo Fisher Scientific, Waltham, MA, USA). An equal amount of proteins from different treatment conditions was used for western blot experiments. The western blot techniques are described in the supplementary materials section. Because the treatments directly affected ACC cell cycles and the doubling time of SW13 and NCI-H295R cells are approximately 24 and 48 h, respectively, the optimal time to capture the changes in molecular signaling was shorter in SW13 (24–48 h) than in NCI-H295R (48–72 h) before ACC cells underwent treatment-related apoptosis.
Cellular migration and invasion assay
To determine the effects of a single drug or combination drugs on the migratory and invasive capacity of ACC cells, we performed cellular migration and invasion assays according to the manufacturer’s instruction (Cat # 354578, Cat # 3544880, BD Bioscience, San Joes, CA, USA). ACC cells were plated in six-well plates in a triplicate manner and treated with varying concentrations of OTS167, RGB-286638, the combination of OTS167 and RGB-286638, and vehicle control for 24 h and 48 h, respectively. The methods of cellular migration and invasion assay are described in the supplementary materials section.
Immunofluorescence analysis
2 × 105 SW13 and NCI-H295R cells were plated on glass coverslips, allowed to attach overnight, and treated for 48 to 72 h. Cells were fixed with 4% paraformaldehyde, permeabilized in 0.25% Triton-X, and incubated overnight with primary antibodies. DNA was stained with DAPI (Vector Laboratories, Burlingame, CA, USA). Images were obtained by fluorescence microscopy with 40 × magnification and collected using Carl Zeiss ZEN Software (Zeiss, Germany).
Separation of polymerized and depolymerized tubulin
Tubulin polymerization and depolymerization assay was performed to check the effects of drug treatments on the tubulin polymerization process in ACC cells. Therefore, SW13 (1 × 106) and NCI-H295R (1 × 106) cells were plated in a 100 mm dish with 10 mL of culture medium. Cells were treated with drug(s) and vehicle control for 48 h for SW13 and 72 h for NCI-H295R cells. The method of separation of polymerized and depolymerized tubulin assays is described in the supplementary materials section.
Oligo small interfering RNA (siRNA)-mediated transfection
SW13 and NCI-H295R cells were transfected with small interfering RNA (siRNA) specific for MELK (4390824, assay ID s386, Thermo Fisher Scientific, MA, USA) or control siRNA (4390843, Thermo Fisher Scientific, MA, USA) using Lipofectamine RNAiMAX (13778–015, Invitrogen; Thermo Fisher Scientific, Inc., MA, USA). After 48 h of transfection, Western Blot was performed to check the transfection efficiency of MELK knockdown and target proteins.
In vivo study
The protocol designed to study the in vivo efficacy of OTS167 and RGB-286638 in mice with human ACC xenografts was approved by the National Cancer Institute, National Institutes of Health (NIH), Animal Care and Use Committee. Mice were maintained according to NIH Animal Research Advisory Committee guidelines. A total of 5 × 106 NCI-H295R cells with luciferase reporter in Corning® Matrigel® Matrix (Cat # 354234, Corning, NY, USA) were injected into each flank of a Nuþ/Nuþ mouse (two xenografts per mouse). After 21 days, mice were randomized into four groups by the treatment. We selected the dosages of OTS167 and RGB-286638 based on prior publications that showed in vivo efficacy with no signs of treatment-related toxicities in mice [16, 17]. Treatments included: Group 1: 0.1% DMSO as vehicle control; Group 2: daily (Monday-Friday) OTS167 (10 mg/kg) via intraperitoneal injection; Group 3: RGB-286638 (20 mg/kg) using an intravenous injection (IV) via tail vein three times (Monday, Wednesday, Friday) weekly for two weeks, followed by RGB-286638 drugs (6 mg/100 μl) loaded ALZET pumps with 0.25 μl/hour delivery rate (Model 1002, Alzet, Cupertino, CA, USA) Group 4: the combination of OTS167 (10 mg/kg) and RGB-286638 (20 mg/kg), following the above protocol for drug administrations. The duration of treatment was five weeks. Mice received daily health monitoring, and mouse weight was recorded weekly. In brief, the details of in vivo imaging studies are described in the supplementary materials.
Statistical analysis
Statistical analyses were performed using SPSS version 25.0 for Windows (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 8 software (GraphPad Software, La Jolla, CA, USA). Statistical analysis methods are described in the supplementary materials section.






留言 (0)