
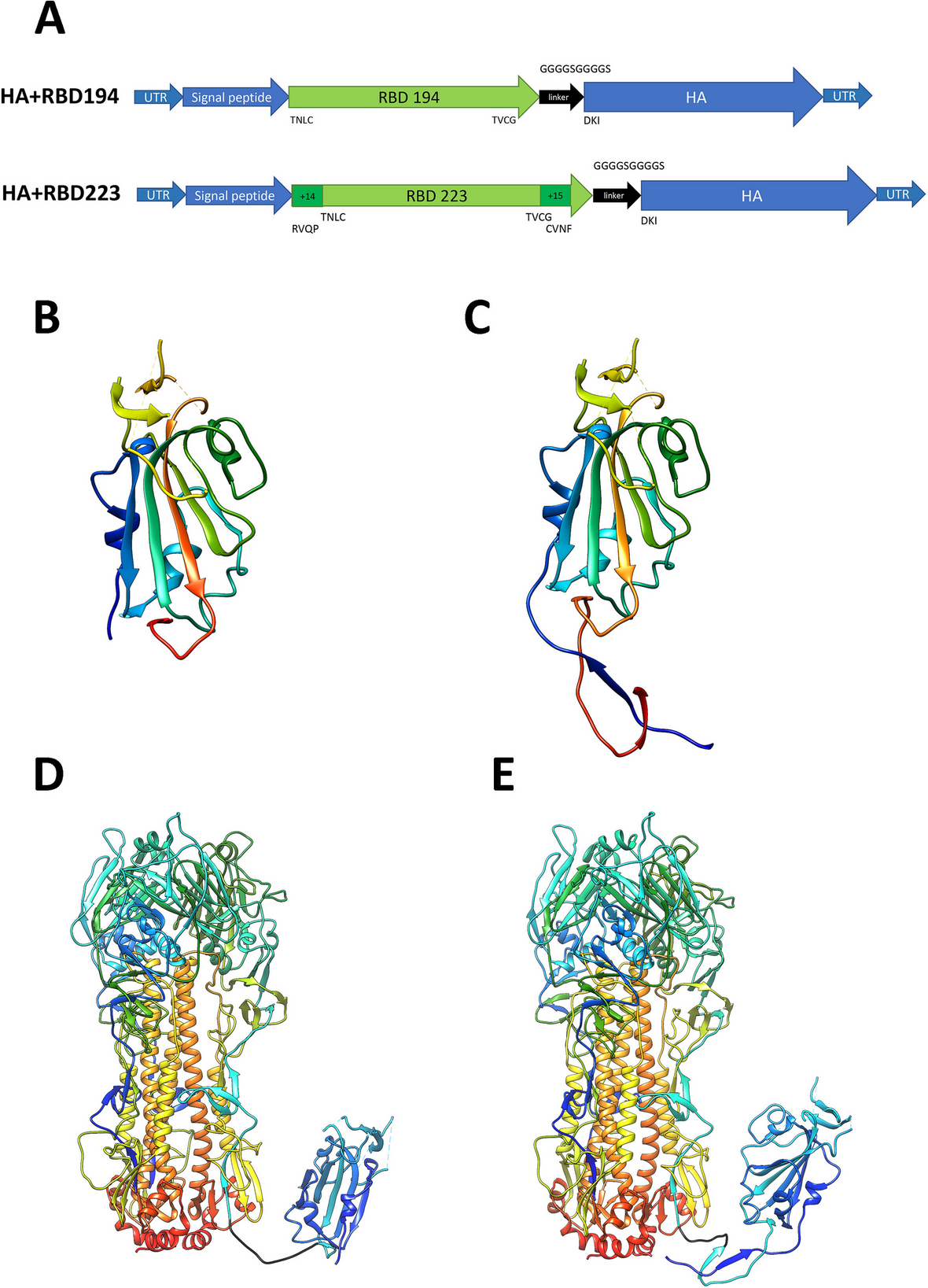
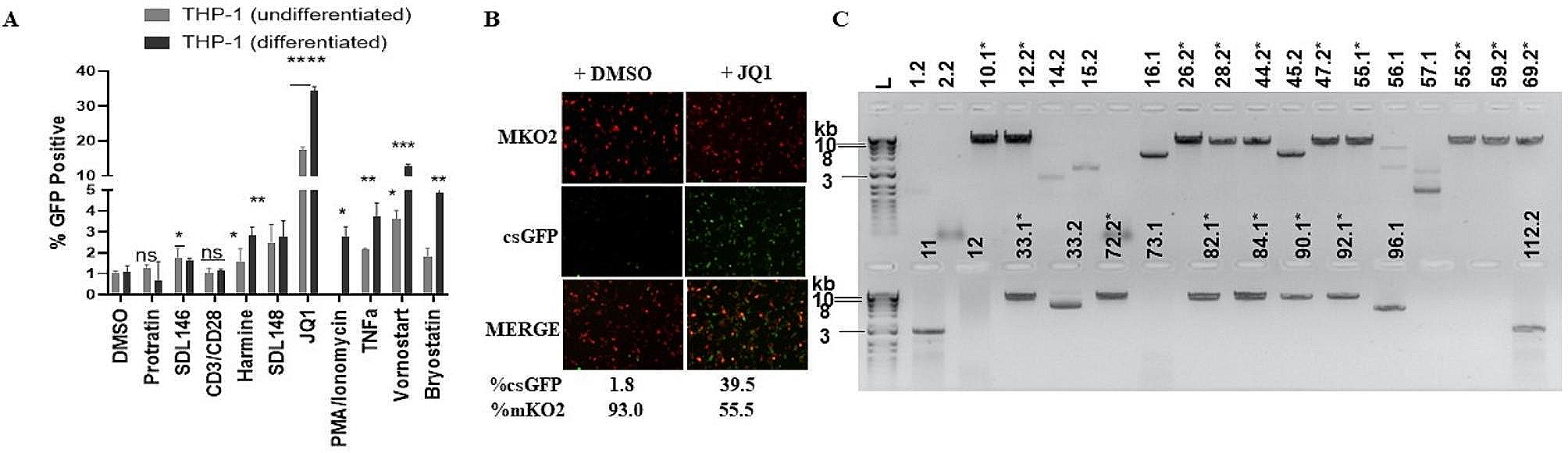
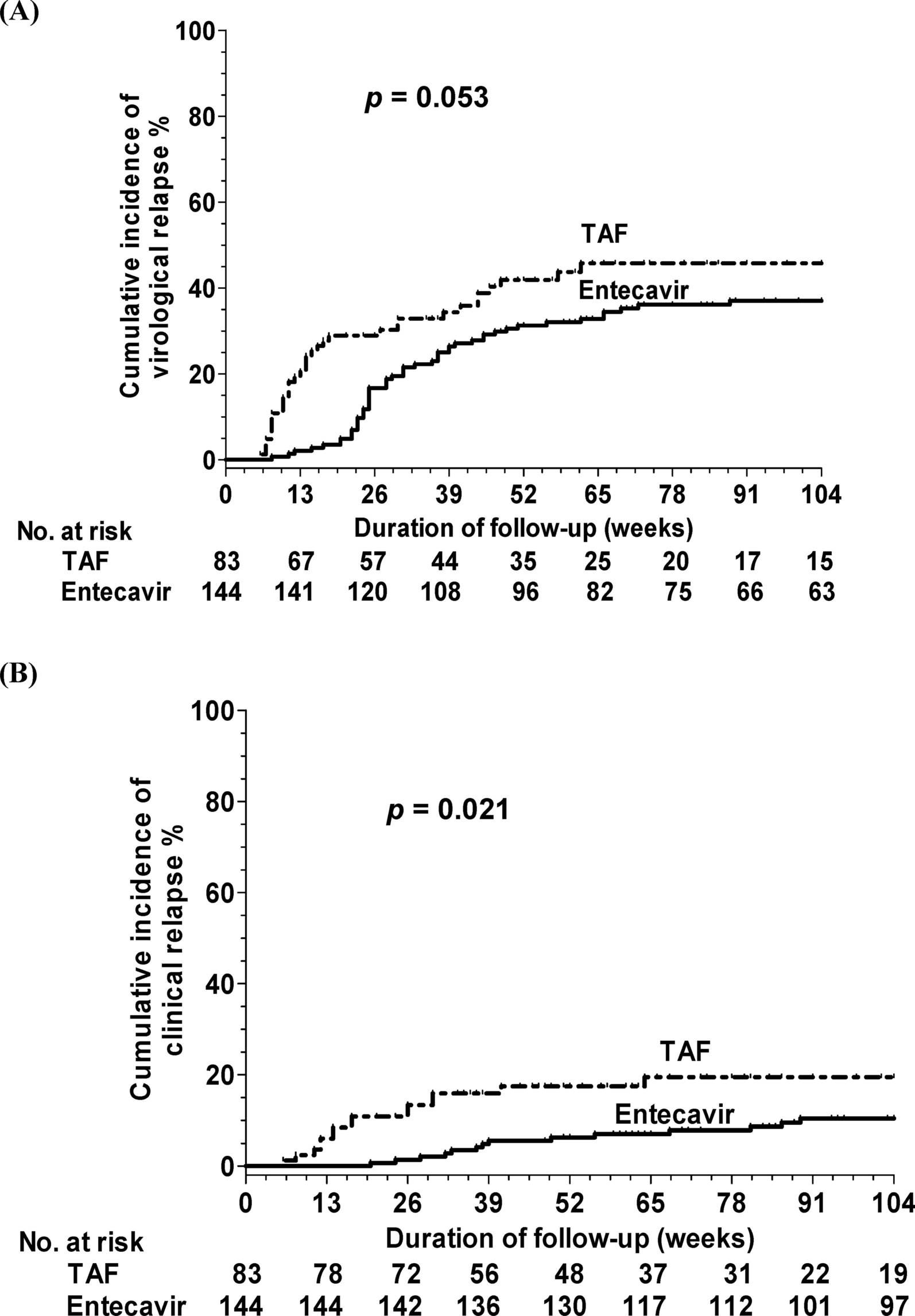
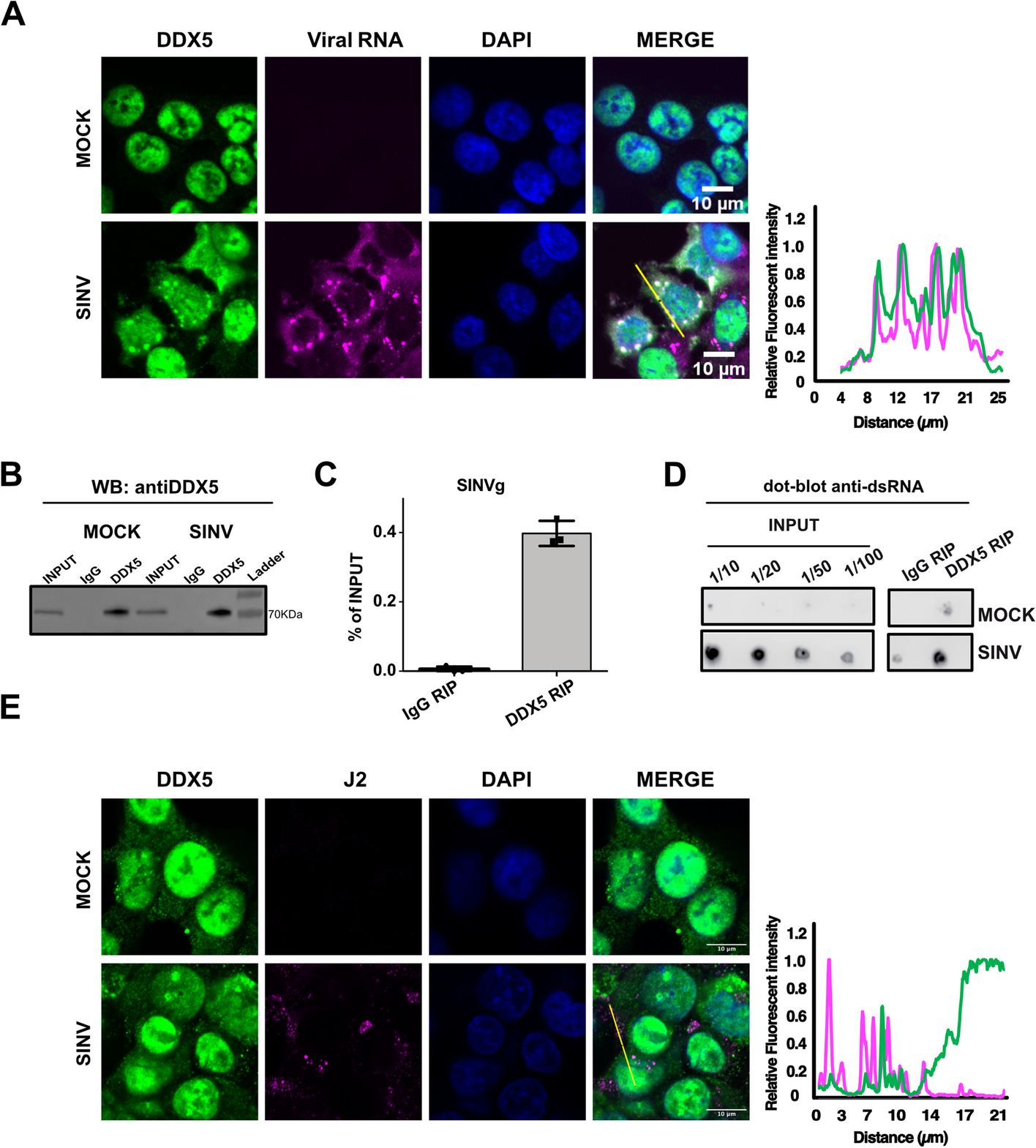
記住我


A total of 80 archived stool specimens with Ct values under 30 (< 30 Ct) were collected from individuals displaying NOV-related clinical symptoms, among which 40 and 30 samples were tested as positive for NOV genotype GII.4 and GII.17, respectively, and 10 samples, including GII. 2, GII. 3 and GII. 6 according to a previous method [22]. These samples used in this study were collected by Ningbo Municipal Center for Disease Control and Prevention (NCDC) between January, 2016 and December, 2020. Ethical approval for this study was obtained from the ethical committee of NCDC.
ReagentsFAM-TTATTATT-quencher (ssDNA FQ), FAM-TTATTATT-biotin (ssDNA FB) and probes were generated by GENEWIZ Inc. (Suzhou, China). Oligonucleotides of all the primers employed in this study were synthesized by Sangon Biotech (Shanghai, China), and displayed in Table 1. RT-RAA nucleic acid amplification kit was purchased from Jiangsu Qitian Gene Biotechnology Co., Ltd (Wuxi, China). NEbuffer 2.1 and Cas12a were purchased from New England Biolabs (MA, USA). The LFS was purchased from Tiosbio (Nanjing, China).
Table 1 The oligonucleotides used for RT-RAA and crRNA in RT-RAA-Cas12a-based assay for norovirus genotype GII.4 and GII.17 detectionProduction of standard RNA of NOVs genotype GII.4 and GII.17Whole genome sequences of different strains of NOVs genotype GII.4 or GII.17 were recovered from the NCBI database and aligned using ClustalW. Based on the alignment result, a specific consensus region of viral protein 1 (VP1) gene corresponding to sequences 4991–5380 was determined as the target (Fig. 1). The DNA fragment of the target sequence was synthesized and inserted into plasmid pBluscript II SK (+) (Sangon, Shanghai, China) (Fig. 1). The resulting pBluscript-NOV was transformed into Escherichia coli TOP10 cells to construct the recombinant strain TOP-NOV. Then, the pBluscript-NOV was extracted using the TIANprep Mini Plasmid Kit (Tiangen Biotech, Beijing, China), and linearized with Sac I. The linear pBluscript-NOV was employed as the template of the in vitro transcription (IVT) reaction to generate NOV RNA standard using IVT T7 Kit (TaKaRa, Dalian, China). The IVT reaction mixture was composed of 5 μL of 10 × transcription buffer, 5 μL of each NTP solution, 1 μL of RNase inhibitor, 5 μL of T7 RNA polymerase, 6.5 μL of RNase-free water and 12.5 μL of linear pBluscript-NOV plasmid, and incubated at 39 °C for 2 h. Finally, RNA concentration in nanogram was converted to that of RNA copy number using the following formula: RNA copy number = [M (ng/μL) × 6.02 × 1023]/(N × 109 × 340), where M refers to the RNA concentration quantified by a spectrophotometer (Metash Instruments, Shanghai, China), N refers to the length of RNA.
Fig. 1
Visualization of primers for reverse transcription recombinase-aided amplification (RT-RAA) and crRNA spacer sites in the target viral protein 1 (VP1) gene sequence of NOV genome. RT-RAA primers are indicated by red rectangles. crRNAs are programmed to specifically target VP1 gene of noroviruses (NOV) GII.4 and GII.17
Design and preparation of crRNAsFour crRNA with different nucleotides of crRNA spacer sequences targeting the highly conserved region of the VP1 gene were determined, and further evaluated for the specificity of each crRNA to NOVs genotype GII.4 or GII.17 using the Basic Local Alignment Search Tool. Moreover, the 21 nucleotides of 5′-TAATTTCTACTAAGTGTAGAT-3′ were used for crRNA stem sequence, and served as a binding scaffold for Cas12a. Preparation of crRNA was performed using the following step. The oligonucleotides for the production of crRNA preparation listed in Table 1 were chemically synthesized by GENEWIZ. (Suzhou, China), and annealed. The resulting double strand DNAs (dsDNA) were further transcribed by in vitro transcription using IVT T7 Kit. The transcription reaction comprised 5 μL of 10 × transcription buffer, 5 μL of each NTP solution, 1 μL of RNase inhibitor, 5 μL of T7 RNA polymerase, 9 μL of RNase-free water and 10 μL of annealed dsDNA, and incubated at 39 °C for 2 h. The resulting crRNA products were purified using phenol–chloroform extraction and isopropanol precipitation, and the concentration of crRNA was determined using a spectrophotometer.
Optimization of reaction condition of RT-RAAThe performance of RT-RAA was evaluated by comparing three factors consisting of different primers, the specificity, and the sensitivity of the assay. The RT-RAA reaction mixture comprised 25 μL of reaction buffer V, 2 μL of each primer (10 μM), 16.5 μL of ddH2O, 2 μL of standard RNA and 2.5 μL of 280 mM magnesium acetate. Then the reaction tubes were placed into the preheated Axxin T8 isothermal instrument for 20 min at 39 °C. Subsequently, the RT-RAA products were treated with an equal volume of 50 μL phenol/chloroform, subjected to agarose gel electrophoresis (2%) and finally evaluated under an ultraviolet (UV) light.
Optimization of RT-RAA-Cas12a-mediated fluorescent and LFS assayThe RT-RAA-Cas12a-mediated assay was carried out using 5 μL of RT-RAA products in a total reaction volume of 50 μL, as well as 45 μL of the CRISPR-Cas12a reaction mixture, which contained 5 μL of 10 × NEBuffer 2.1, 1 μL of 100 nM crRNA, 1 μL of 1 μM Cas12a, 0.5 μL RNase inhibitor (40 U), and 2 μL of 1 μM ssDNA reporters, 35.5 μL of RNase-free water. Then, the reactions were conducted in the preheated Axxin T8 isothermal instrument for 15 min at 39 °C with fluorescent signals collected every 10 s (ssDNA FQ substrates = λex: 485 nm; λem: 535 nm), and visualized by a UV light illuminator and LFS with ssDNA FB as the substrates.
Specificity of RT-RAA-Cas12a-mediated fluorescent and LFS assayThe specificity of RT-RAA-Cas12a-mediated assay was performed by testing each RNA samples at a concentration of 200 copies/μL from other gastrointestinal viruses, including human rotavirus (HRV), astrovirus (HAtV), and enterovirus 71 (HEV71). 10 different samples testing positive for each test virus as well as 10 non-GII.4 or GII.17 samples were employed as the control in this study.
Sensitivity of RT-RAA-Cas12a-mediated fluorescent and LFS assayTo evaluate the sensitivity of RT-RAA-Cas12a-mediated assay, a concentration gradient of RNA standard samples, which was adjusted to be 200, 100, 10, 1, 0.5, 0.1 and 0.05 copies/µL, was used as the template in RT-RAA reaction. All volumes of Cas12a-mediated assay were 50 µL as described above. Each reaction process was replicated three times and the results were analyzed by fluorescence and LFS.
RT-PCR for clinical samplesThe total RNA of 80 samples, each 200 µL of clinical solution, was manually extracted using BeaverBeads™ Viral DNA/RNA Kit BEAVER, Suzhou, China), and automatically performed using automatic nucleic acid extraction instrument (bioPerfectus technologies, Jiangsu, China), respectively. The RT-PCR detection for NOV nucleic acids of clinical samples was performed using NOV test kit (bioPerfectus technologies, Jiangsu, China) in an ABI 7500 (Applied Biosystems). The reactions were performed with an initial step of reverse transcription at 48 °C for 30 min, followed by 95 °C for 15 s, 35 cycles of 95 °C for 15 s, and 53 °C for 1 min.
StatisticsEach sample was conducted in at least three independent biological replicates. Statistical analysis of the data involving end-point fluorescence was performed using the Prism 8 (GraphPad Software, version 8.0.1) and statistical differences were assessed by the Students’ t-test. The unpaired twotailed t-test was applied to investigate the differences between groups and the threshold for defining significance was based on the p value < 0.05.
留言 (0)