
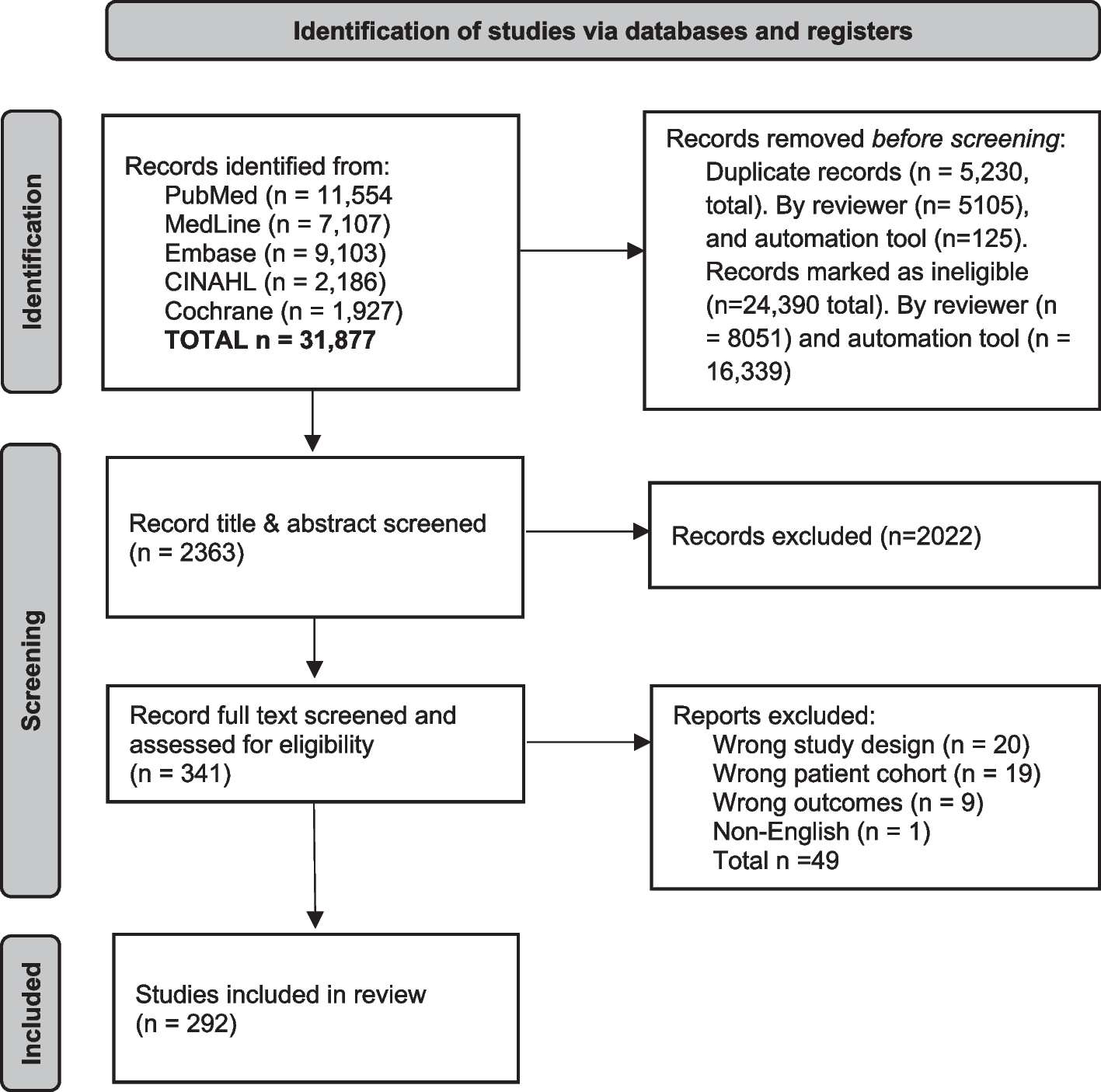
記住我

To assess the distribution of genetic alterations (e.g. inframe mutation, missense mutation, nonsense mutation, fusion, amplification, and nonstop mutation) in PSM genes in different cancer types, we used genomic profiling data retrieved from the web-based cBioPortal tool for over 10,000 tumor samples (representing 33 cancer types and 11 pan-cancer body groups) from the TCGA dataset (Tables 1 and 2). PSM genes were shown to be altered in approximately 67% of esophageal carcinoma (ESCA) cases (n = 182) and 66% of lung squamous cell carcinomas (LUSC, n = 487), but only 4% of thyroid carcinoma (THCA) cases (n = 500; Fig. 2A). Genetic alterations (predominantly DNA amplification) were subsequently detected in all PSM genes, with the vast majority of aberrations found in the PSMD2 (6% of patient samples), PSMB4 (4%), and PSMD4 (4%) genes. In contrast, relatively few samples were found to harbor mutations in the PSMA3 gene (approximately 1%; Supplementary Fig. 1). Interestingly, genetic aberrations in PSMD2 were most frequently found in LUSC (37% of 487 cases).
Fig. 2
Bar charts depicting alteration frequency for the 49 PSM genes by cancer type using the interactive web-based online tool cBioPortal (cbioportal.org). A DNA amplification was shown to be prevalent in most cancer types, with ESCA and THCA showing the highest och lowest alteration frequencies, respectively. Box plots visualizing DNA amplification of (B) PSMB3, (C) PSMB4, (D) PSMD4 and their effect on expression (RSEM). Wilcoxon test was used to calculate statistical significance (Benjamini–Hochberg adjusted p-values), ns = not significant (P ≥ 0.05); *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. E PSME4 gene was the most mutated of all PSM genes. Most PSME4 mutations was found in the UCEC cancer type, where missense mutations were prevalent. F Beeswarm plot visualizing copy number alterations (CNA) and other types of mutations, and their effect on expression was generated in cBioPortal. Deep deletions in PSME4 resulted in significantly lower expression. G Lollipop plot depicting the number of mutations across the PSME4 gene. Missense mutations were prevalent (243 of 312 mutations), with a domain with unknown function containing 14 mutations (10 frameshift deletions in T1805Pfs*69, three frameshift insertions in T1805Nfs*11, and one missense in T1805P)
GISTIC2 data from Broad GDAC Firehose were then used to evaluate the effect of DNA amplification of the 49 PSM genes on gene expression (Supplementary Table 1). Broad amplification of whole chromosome arms (p and q arms) was most prevalent in the different cancer types (mean ± SEM, 7.3 ± 0.9; range, 1–22), while focal amplification was found in 1.7 ± 0.4 (range, 0–12) cancer types per PSM gene. Furthermore, similar DNA amplification profiles were found for 10 PSM genes located on the same cytoband (PSMB5 and PSMB11, 14q11.2; PSME1 and PSME2, 14q12; PSMC4 and PSMD8, 19q13.2; PSMB4 and PSMD4, 1q21.3; PSMB8 and PSMB9, 6p21.32; Supplementary Fig. 2) and a number of consensus cancer driver genes (e.g. PSMB3 and ERBB2, 17q12; PSME3 and BRCA1, 17q21.31) [43, 44]. Moreover, several PSM genes (PSMA6-8, PSMB3-4, PSMB8-9, PSMC2, PSMC4-5, PSMD2-4, PSMD8, PSMD12, and PSMG3-4) were amplified > 100 times across cancer types. Of these, PSMB4 (1q21.3) and PSMD4 (1q21.3) genes were amplified > 400 times, while PSMD2 (3q27.1) was amplified almost 600 times. In general, DNA amplification was most prevalent in the BLCA (urologic), BRCA (gynecologic), LUSC (thoracic), LUAD (thoracic), OV (gynecologic), and UCEC (gynecologic) cancer types. DNA amplification events (broad and focal) resulted in significantly elevated RNA levels for all 49 PSM genes in amplified samples compared to non-amplified samples (P adjusted < 0.05; Supplementary Table 1), including PSMB4 (1q21.3), PSMD4 (1q21.3), and PSMB3 (17q12) that demonstrated focal amplifications in > 10 cancer types (Fig. 2B-D).
In total, 3% of the 2,935 genetic variants were found to harbor DNA amplification of PSM genes (n = 31) in conjunction with mutations (n = 37; BLCA, BRCA, CESC, COADREAD, ESCA, HNSC, LUAD, LUSC, SARC, SKCM, STAD, UCEC) or fusions (n = 40; BLCA, BRCA, CESC, CHOL, ESCA, LIHC, LUAD, OV, SARC, SKCM, UCS) in the same patient (Supplementary Tables 1 and 4). Although all 77 co-occurrences of amplification/mutation or amplification/fusion were unique, six patients with BRCA, CHOL, HNSC, LIHC, LUAD, or UCEC harbored two different amplification/mutation (PSMC2 or PSMC5) or amplification/fusion events (PSMB2 or PSMD11) in the same gene or two different genes (PSMD4 and PSMG3 in a LUAD sample, and PSMD11 and PSMD12 in a BRCA sample). The PSM gene was most commonly the 5’- gene partner (58%), and co-expression between the fusion gene partners was relatively weak (rs <|0.4|). According to Polyphen-2 functional prediction annotation scores, 18/40 amplification/fusion and 17/37 amplification/mutation events were predicted to be possibly damaging (Polyphen-2 scores 0.15 to 1). In contrast, 12/40 amplification/fusion events in PSMB2, PSMB3, PSMC4, PSMD3, PSMD4, and PSMD11, and 12/37 amplification/mutation events in PSMA6, PSMA8, PSMB8, PSMC2, PSMC6, PSMD2, PSMD3, and PSMD4 were more confidently predicted to be damaging (Polyphen-2 scores 0.85 to 1).
Of the 2,935 genetic variants identified in the 49 PSM genes, 2,782 (95%) were classified as potentially deleterious (Supplementary Table 4). Although SIFT and/or Polyphen-2 functional prediction annotation data were not available for 1,233 of the 2,782 (44%) genetic variants, 961 and 900 potentially damaging variants were identified, respectively. Consequently, 721 potentially damaging variants were identified by both databases in 28/32 cancer types and in all PSM genes, except PSMB10 and PSMG1-4. Of the 49 PSM genes, PSME4 had the highest number of mutations, primarily consisting of missense mutations though other mutations were also identified (e.g. nonsense mutation, fusions, amplifications; Fig. 2E). As expected, copy number alterations in the PSME4 gene such as amplification and deep deletion resulted in over- and underexpression, respectively. However, PSME4 expression varied in samples harboring missense mutations (Fig. 2F). Although missense mutations spanned the PSME4 gene, 14 cancer samples (colon adenocarcinoma (COAD, n = 2), stomach adenocarcinoma (STAD, n = 6), and uterine corpus endometrial carcinoma (UCEC, n = 6)) had truncating mutations in a domain at the C-terminal region with unknown function (10 with frameshift deletion in T1805Pfs*69, three with frameshift insertion in T1805Nfs*11, and one sample with missense in T1805P; Fig. 2G).
In the breast cancer validation dataset, only PSMA4 (HER2/ER- subtype, n = 2; bilateral breast cancer), PSMB7 (Luminal B/HER2- subtype, n = 1), PSMD3 (Luminal B/HER2- subtype, n = 3; Luminal B/HER2 + subtype, n = 1; Basal-like subtype, n = 1), and PSME4 (Luminal B/HER2- subtype, n = 2) harbored mutations. DNA amplification was prevalent in 33/39 PSM genes, where five genes (PSMA7, PSMB4, PSMD2-4, PSMD10) were amplified in more than 10% of all samples (Supplementary Table 3). These five genes were significantly overexpressed in amplified samples compared to non-amplified breast cancer samples (P < 0.0001; t-test). Amplification of PSMA7, PSMB4, PSMD4, and PSMD10 were identified in the Luminal B, HER2/ER-, and Basal-like subtypes, while PSMD3 amplification was only found in Luminal B and HER2/ER- samples and PSMD2 amplification in Luminal B and Basal-like samples. These findings were in agreement with the cBioPortal TCGA dataset. Taken together, these data show that although genetic aberrations were found in all PSM genes, specific PSM genes are hotspots for DNA amplification in certain cancer types.
Differential gene expression analysis between cancer and normal tissues identifies cancer-related PSM genesDifferential gene expression analysis was performed in 16/33 cancer types using RNA-seq data from TCGA cancer samples (n = 5,507) with corresponding normal tissue (n = 627). Expression profiling of 49 PSM genes revealed similar gene expression patterns across the different cancer types, frequently showing overexpression in cancer in comparison with normal tissue (Fig. 3). Interestingly, hierarchical clustering revealed two main clusters of PSM genes, of which one cluster contained five PSM genes (PSMB8-10 and PSME1-2) with high expression in a number of urologic, CNS, and gynecological cancers (Fig. 3). Furthermore, differential expression was found in 35 ± 2 (mean ± SEM, range 17–45) PSM genes per cancer type. Interestingly, 45/49 PSM genes were differentially expressed in the breast invasive carcinoma (BRCA) and lung squamous cell carcinoma (LUSC) cancer types, while only 17/49 PSM genes were differentially expressed in pheochromocytoma and paraganglioma (PCPG; Fig. 4A). Moreover, 11 ± 0.4 (range 2–15) cancer types were associated with each PSM gene. Overexpression of PSM genes was most prevalent across the range of cancer types. For instance, seven PSM genes (i.e. PSMA1, PSMA4, PSMC1, PSMC3IP, PSMD13, PSMG2-3 (PSM class I/II/V)) were overexpressed in the majority of the 16 cancer types (Fig. 4B). In comparison with the other PSM genes, differential expression of PSMB11 was relatively uncommon, whereas PSME3 and PSMG3 were found to be differentially expressed in virtually all examined cancer forms (15/16 cancer types; Fig. 4C-D). Taken together, these findings demonstrate that the vast majority of PSM genes were cancer-related.
Fig. 3
Human proteasome genes frequently displayed overexpression in cancer compared with normal tissue. Heatmap showing relative log2 RSEM gene expression (cancer vs mean normal samples) for the 49 PSM genes in 5,507 TCGA cancer samples representing 16 pan-cancer diseases. Hierarchical clustering was performed with the pheatmap R package (version 1.0.12) using the Manhattan distance metric and Ward’s minimum variance method (Ward.D2)
Fig. 4
Differentially expressed PSMs between 16 cancer types and corresponding normal tissue. A Bar chart visualizing the number of differentially expressed PSM genes between cancer and normal tissue. BRCA and LUSC showed the highest number of cancer-related PSMs (n = 45), whereas only 17 differentially expressed PSMs were identified in PCPG. B Bar chart depicting differential PSM gene expression patterns in various cancer types. Overexpression strongly dominated across all cancer types. C-D Box plot depicting differentially expressed PSMs in cancer and normal tissue. PSMB11 was found to be differentially expressed in 2/16 cancer types, while PSME3 was differentially expressed in all except one of the 16 cancer types. The Wilcoxon test was used to calculate statistical significance (Benjamini–Hochberg adjusted p-values) differences in expression (RSEM) between cancer and normal tissue. ns = not significant (P > 0.05); *P < 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001
Pearson correlation reveals five clusters of co-expressed PSM genes in cancerTo assess co-expression of the 49 PSM genes in cancer, pairwise Pearson correlation coefficients (r) were calculated for the PSM genes in the 33 cancer types. First, we evaluated overall PSM co-expression patterns in cancer by compiling RNA-seq data for all 33 cancer types. This analysis showed that the majority of co-expressed PSM genes were positively correlated, with at least five gene clusters displaying moderate to strong positive correlation (r >|0.4|: 1) PSMD1, PSMD11-12, PSME3-4, 2) PSMA3-4, PSMA6, PSMC6, 3) PSMA2, PSMA5, PSMA7, PSMB2, 4) PSMB1, PSMB3-7, PSMC1, PSMC3, PSMC5, PSMD4, PSMD9, PSMD13, PSMG3, and 5) PSMB8-10, PSME1-2; Fig. 5A). In contrast, Pearson correlation coefficients varied between |0.4| and |0.9| for the 33 cancer types. Interestingly, PSMB8-10 (PSM class I) displayed moderate to strong positive correlation patterns in 31 cancer types (e.g. KIRC, LIHC, LUAD). Furthermore, PSMB8-10 (PSM class I) expression was also strongly correlated with PSME1-2 (PSM class III) in 27 cancer types, e.g. BRCA (Fig. 5B). Consequently, a number of PSM genes belonging to different PSM gene classes were found to be positively correlated, particularly PSMB8-10, which are found in the immunoproteasome.
Fig. 5
Pairwise Pearson correlation between PSM gene expression in 33 pan-cancer diseases. Correlation matrices for compiled gene expression patterns for (A) the 33 pan-cancer diseases and (B) BRCA, with genes ordered using hierarchical clustering with Ward’s minimum variance (Ward.D2). Red and blue dots represent negative and positive correlation patterns, respectively. The strength of color and circle size defines correlation pattern between gene pairs using correlation coefficients (P < 0.05); blank squares were not statistically significant (P > 0.05). PSM genes showing recurrent positive correlation are outlined in red
Multivariable Cox regression analysis shows the prognostic significance of PSM gene expression in cancerTo assess the prognostic significance of PSM genes, log2 Fragments Per Kilobase of transcript per Million (FPKM) gene expression (RNA-seq) values were retrieved from the web-based UCSC Xena Browser tool for 10,304 GDC TCGA samples (representing 33 cancer types and 11 pan-cancer body groups; Table 2). Survival analysis was then performed to evaluate the prognostic relevance of the 49 PSM genes in 33 cancer types using overall survival (OS) and progression-free interval (PFI) as clinical endpoints adjusted for covariates (age for 33 cancer types and/or tumor grade for 12 cancer types; Fig. 6A-B). Survival analysis for PFI could not be performed for acute myeloid leukemia (LAML) due to a lack of clinical data. In total, age was shown to have an adverse effect on OS in 22/33 cancer types (e.g. BRCA, OV, and UVM) and 5/32 cancer types (e.g. CESC, LGG, and SKCM) for PFI, but tumor grade only affected prognosis in 3/12 cancer types (i.e. HNSC, PAAD, and UCEC) for OS and 4/12 (e.g. ESCA, KIRC, and PAAD) for PFI.
Fig. 6
The prognostic relevance of PSM gene expression in different cancer types using overall survival (OS) and progression-free interval (PFI) as clinical endpoints in multivariable Cox regression analysis (adjusted for age and/or tumor grade). A-B Dot plots displaying the –log10(p-value) for the multivariable Cox regression analysis between PSM gene expression and OS (A) and PFI (B). Blue dots indicate a hazardous role for PSM gene expression, while red dots indicate a protective role. NS = not significant (P > 0.05). Dot sizes denote –log10(p-value); P < 0.001 is shown as –log10(p-value) = 3. Due to a lack of clinical data, PFI could not be performed for acute myeloid leukemia (LAML). C-D Bar charts illustrating the number of cancer types associated with different expression levels for each prognostic PSM gene. PSM gene expression (high [blue bars, higher than median expression] and low [yellow bars, lower than median expression]) associated with OS (C) and PFI (D) in different cancer types
In total, PSM gene expression (high or low expression) was shown to affect prognosis in 7.1 ± 0.4 (mean ± SEM, range 2–14 (OS)) and 6.0 ± 0.3 (mean ± SEM, range, 2–11 (PFI)) cancer types (Fig. 6C-D and Supplementary Fig. 3). Furthermore, PSM genes linked to decreased survival (OS and PFI) were also investigated in ≥ 30% of cancer types. For OS, 12 prognostic PSM genes (i.e. PSMA1, PSMA4, PSMB4-5, PSMB8, PSMB10, PSMD2, PSMD11-12, PSMD14, PSME2, and PSMG1; PSM class I/II/III/V) were identified in ≥ 30% of cancer types (Fig. 6C), whereas only two PSM genes (PSMA1, PSMD11; PSM class I/II) were identified for PFI (Fig. 6D). In addition, PSMD2 had an impact on prognosis in 42% (14/33) of all cancer types for OS (Supplementary Fig. 4). Interestingly, PSMB8-10 and PSME1-2 genes had a significant impact on OS in most cancer types, primarily when underexpressed (Fig. 6C). In contrast, overexpression of PSMB5, an important catalytic site in the proteasome, was associated with decreased OS and PFI in 36% and 27% of cancer types, respectively (Figs. 6C-D and 7A-B).
Fig. 7
The number of prognostic PSMs associated with high or low expression per cancer type using overall survival (OS) and progression-free interval (PFI) as clinical endpoints in multivariable Cox regression analysis (adjusted for age and/or tumor grade). A-B Forest plots visualizing the Hazard ratio (HR) for the multivariable Cox regression analysis between high PSMB5 expression and OS (A) and PFI (B). HR < 1 shows reduced risk at high PSMB5 expression (higher than median expression) and HR > 1 illustrates increased risk at high PSMB5 expression. C-D Bar charts visualizing the number of prognostic PSMs associated with each cancer type at high (blue bars, higher than median expression) or low (yellow bars, lower than median expression) expression for OS (C) and PFI (D)
In contrast, specific cancer types were associated with 10.6 ± 1.6 (range, 0–31 (OS)) and 9.0 ± 1.6 (range, 0–31 (PFI)) prognostic PSM genes (Fig. 7C-D). Moreover, specific cancer types were identified where ≥ 50% of PSM genes (up- or downregulation) were linked to more unfavorable survival, with overexpression being most common. For OS, four cancer types (i.e. ACC (29 genes), LGG (26 genes), LIHC (26 genes), and UVM (31 genes)) were identified (Fig. 7C), and three cancer types (i.e. ACC (29 genes), KIRP (25 genes), and UVM (31 genes)) were identified for PFI (Fig. 7D). Interestingly, > 60% of PSM genes (predominantly overexpressed) were associated with both reduced OS and PFI in UVM (Fig. 7C-D and Supplementary Fig. 4). Consequently, these results show that PSM gene expression patterns may be an important indicator of prognosis in various cancer types. Compared to the TCGA dataset, similar correlation patterns between PSM gene expression and survival were observed in the breast cancer validation dataset and KM plotter (Supplementary Table 5).
留言 (0)