Explanation for the choice of comparators
The ideal blood glucose target remains debated, and local blood glucose control practices vary widely [21,22,23,24]. In the control group of the TGC-fast RCT (liberal blood glucose control), hyperglycemia is only treated when above 215 mg/dl (11.9 mmol/l) and hence exceeding the renal threshold, above which obvious complications may ensue [16]. This threshold to initiate insulin treatment in the control group was also used in the pioneer RCTs investigating tight versus liberal blood glucose control [5, 8, 9].
In both study groups, blood glucose concentrations are measured by an on-site blood gas analyzer in undiluted blood drawn from the arterial line. Glucose measurement on arterial blood using an on-site blood gas analyzer yields both a fast and accurate measurement of blood glucose in critically ill patients [14]. When the arterial catheter is no longer needed (for medical reasons), blood glucose is measured on capillary blood using a glucometer with a validated performance. Measurement of the blood glucose concentration on blood drawn from the (central) venous catheter through which insulin and glucose are administered is not allowed due to potential interference with the measurement. In both study groups, insulin is only infused through continuous intravenous infusion through a central venous catheter by a syringe pump, usually in concentrations of 50 IU in 50 mL 0.9% NaCl. The concentration can be increased to 100 IU in 50 mL NaCl 0.9% in case of a high insulin need. No boluses of insulin are allowed.
In the liberal glucose control group (control group), insulin is initiated when blood glucose concentrations exceed 215 mg/dl (11.9 mmol/l) on two consecutive measurements, with the dose adjusted by the nurses/physicians to maintain concentrations between 180 and 215 mg/dl (10–11.9 mmol/l). When blood glucose drops below 180 mg/dl (10 mmol/l), insulin infusion is stopped (except for type 1 diabetics). In type 1 diabetics, insulin is initiated after the first blood glucose measurement above 215 mg/dl (11.9 mmol/l) and the infusion rate is adjusted to maintain blood glucose concentrations between 180 and 215 mg/dl (between 10 and 11.9 mmol/l). Blood glucose is measured minimum 4 times per day. The management of eventual hypoglycemia is at the discretion of the attending physician. To improve protocol compliance, an advisory alert tool was developed and integrated in the patient data management system. This alert advises on whether to initiate/continue or stop insulin administration, without giving advice on the dose. Hence, when blood glucose exceeds 215 mg/dl (11.9 mmol/l) on two consecutive measurements with 4-h interval (or one measurement in type 1 diabetics), the alert indicates to administer insulin; when it drops below 180 mg/dl (10 mmol/l), the alert advises to stop insulin administration (or at least taper down in type 1 diabetics). In addition, the alert advises to measure blood glucose again at the latest after 6 h. The nurse/physician is able to overrule the given advice, e.g., to stop insulin when blood glucose concentrations would be above 215 mg/dl (11.9 mmol/l) but the concentrations are dropping rapidly.
Intervention description
In the tight blood glucose control group, insulin is administered to target the normal healthy fasting ranges for blood glucose (80–110 mg/dl, 4.4–6.1 mmol/l). Insulin is started as soon as blood glucose exceeds the upper normal limit (110 mg/dl, 4.4 mmol/l). Tight glucose control is guided by the LOGIC-Insulin computerized algorithm [19, 20]. The LOGIC-Insulin software advises the nurse on the insulin dosage (or a dextrose bolus in case of hypoglycemia) as well as on the next blood sampling interval. The software was previously validated in a multicenter trial, demonstrating efficacy and safety [20]. The algorithm takes into account the patient profile, (changes) in nutritional intake, the use of drugs such as steroids, and the trend in blood glucose concentrations and insulin dose. The advised sampling interval varies from 1 to 4 h (and more frequent after hypoglycemia), depending on the (observed and predicted) blood glucose stability. Visual alarms on sampling time, hypoglycemia, and nutrition dose entry errors are included in the software. The software is run from a central server in the hospital onto the client bedside computer. The nurses in charge of the patient operate the program. Because the LOGIC-Insulin software serves as an advising system, the nurse or physician has the ability to overrule the given advice.
Criteria for discontinuing or modifying allocated interventions
Tight glucose control is discontinued when the patient starts oral intake of carbohydrates, when the central venous catheter is removed, or at discharge to the general ward or to another ICU not participating in the trial. Eventually, when a patient would stop the oral intake of carbohydrates again while still in ICU, or when the central venous catheter would be replaced in ICU, the intervention is resumed. Upon discontinuation of the study intervention, conventional blood glucose management is applied, which may slightly differ per center, but in general signifies a liberal blood glucose management.
Also in the control group, the alert tool is used until the patient starts oral intake of carbohydrates, when the central venous catheter is removed, or at discharge to the general ward or to another ICU not participating in the trial. At that time, conventional blood glucose management is applied, which may slightly differ per center, but in general signifies a liberal blood glucose management. Eventually, when a patient would stop the oral intake of carbohydrates again while still in ICU, or when the central venous catheter would be replaced in ICU, the alert tool is used again.
Strategies to improve adherence to interventions
The compliance to the allocated study protocol is monitored daily by the clinical trial assistants (CTAs). In both groups, adherence to the protocol is facilitated by the use of decision-support software. In the tight glucose control group, the LOGIC-Insulin software is used, which was previously validated and which advises the nurse on the insulin dosage (or a dextrose bolus in case of hypoglycemia) as well as on the next blood sampling interval [20]. In the liberal glucose control group, an advisory alert tool is used that advises on whether to initiate, continue, or stop insulin administration. The alert also advises to measure blood glucose again at the latest after 6 h.
Relevant concomitant care permitted or prohibited during the trial
In accordance with the recent feeding guidelines for critically ill patients, enteral nutrition is started as soon as possible [25]. When enteral nutrition is insufficient to meet the caloric requirements, supplemental parenteral nutrition is not initiated before day 8 in ICU, in accordance with recent evidence [17, 18]. Except from a small amount of parenteral glucose, no other macronutrients are administered by the parenteral route before day 8. Only in the most severely malnourished patients (body mass index below 17 kg/m2) and in patients readmitted to the ICU, parenteral nutrition can be initiated earlier, as these patients were excluded from large feeding RCTs [17]. In case early parenteral nutrition would be planned upon ICU admission in such patient, the patient is not included in the trial. In general, the amount of parenteral glucose given during the first week in ICU by maintenance solutions may not exceed the equivalent amount of 1 ml dextrose 5% per kg per hour, unless the patient develops spontaneous hypoglycemia (hypoglycemia while not on insulin treatment) or has high risk to do so (e.g., in acute liver failure), or when the patient has a need for high volumes of hypotonic fluids (e.g., severe hypernatremia due to fluid losses). Until the patient receives at least 80% of his/her caloric needs of enteral nutrition, micronutrients (trace elements, minerals and vitamins) are administered parenterally to prevent refeeding syndrome, according to local standard practice.
Provisions for post-trial care
When the RCT would show efficacy and safety of one of two studied treatment strategies, this treatment strategy will be applied to all ICU patients of participating centers.
In accordance with the Belgian and European legislation, the sponsor has a no fault insurance that covers any damage incurred by a study patient and linked directly or indirectly to the participation to the study.
Outcomes
To provide insight in the quality of glucose control in both groups, blood glucose metrics in ICU will be reported, as mentioned in the protocol (see Additional file 1), such as the peak and mean glucose concentrations in ICU, and the incidence of moderate (40–70 mg/dl, 2.2–3.9 mmol/l) and severe (<40 mg/dl; <2.2 mmol/l) hypoglycemia during ICU stay.
The primary outcome is the duration of dependency on intensive care. The duration of dependency on intensive care will be reported as the crude number of ICU-stay days and as the time to live discharge from ICU, to account for mortality as competing risk. ICU non-survivors will be censored beyond the longest duration of ICU length of stay of the survivors. As the timing of ICU discharge to a regular ward may be affected by the availability of beds on regular wards, which could induce bias, we decided to analyze “time to discharge from ICU” as “time to ready for discharge from ICU.” A patient is considered “ready for discharge” as soon as all clinical conditions for ICU discharge have been fulfilled (no longer in need for, or at risk of, vital organ support).
Safety endpoints include:
Secondary efficacy endpoints include:
Hospital length of stay and time to (live) discharge from hospital
Time to final (live) weaning from mechanical respiratory support and the need for tracheostomy
The incidence and type of new infections during ICU stay, and the duration of antibiotic treatment in ICU
Markers of inflammation, including peak values and time profiles
Presence of clinical, electrophysiological, morphological, and molecular signs of respiratory and peripheral muscle weakness during ICU stay
New kidney injury during ICU stay: the presence or absence and duration of new kidney injury during ICU according to KDIGO criteria (Kidney Disease: Improving Global Outcomes) [26, 27]; proportion of patients in need of new initiation of renal replacement therapy in ICU; duration of renal replacement therapy in ICU; recovery from kidney injury and from renal replacement therapy.
The need for pharmacological or mechanical hemodynamic support during ICU stay and its duration, and the time to final (live) weaning from all pharmacological or mechanical support
The incidence and recurrence of atrial fibrillation during ICU stay, duration of atrial fibrillation, number of episodes of atrial fibrillation and treatment for atrial fibrillation (in selected centers)
The incidence of major adverse cardiovascular events (non-fatal myocardial infarction, non-fatal stroke, low cardiac output syndrome, and cardiovascular death) during ICU stay (in selected centers)
The time course of markers of liver dysfunction in ICU, including markers of cholestatic and cytolytic liver dysfunction
The number of readmissions to the ICU within 48 h after discharge
The presence or absence of delirium during ICU stay (in selected centers)
Long-term functional outcome:
◦ For all patients: a validated health questionnaire (Short Form 36, SF-36) 2 years ± 2 months after inclusion.
◦ Subgroup of brain-injured patients (i.e., patients admitted because of traumatic brain injury, subarachnoid hemorrhage, intracranial bleeding, ischemic stroke, or out-of-hospital cardiac arrest): additional functional outcome after 6(±1) and 12(±1) months (extended Glasgow outcome scale and modified Rankin scale)
Use of intensive care resources (costs for hospitalization, for honoraria for medical and allied healthcare services, for pharmacy, for blood products, for clinical chemistry, for radiology, for graft products and for other expenses)
Depending on additional funding, further preplanned studies, of which the detailed protocols will be reported separately, include:
Muscle strength, rehabilitation, recovery of organ function and survival up to 4 years post randomization
Mechanistic studies. The effect of the intervention will be studied in relation to outcome on biochemical, metabolic, immunological, endocrine, inflammatory, coagulation, cardiac and (epi)genetic markers on blood, pleural/pericardial fluid, urine and tissue samples up to 4 years post randomization. Markers include, among others, glucose, lipid, ketone and amino acid concentrations, cytokines, hypothalamic-pituitary hormones, glucagon, and C-peptide.
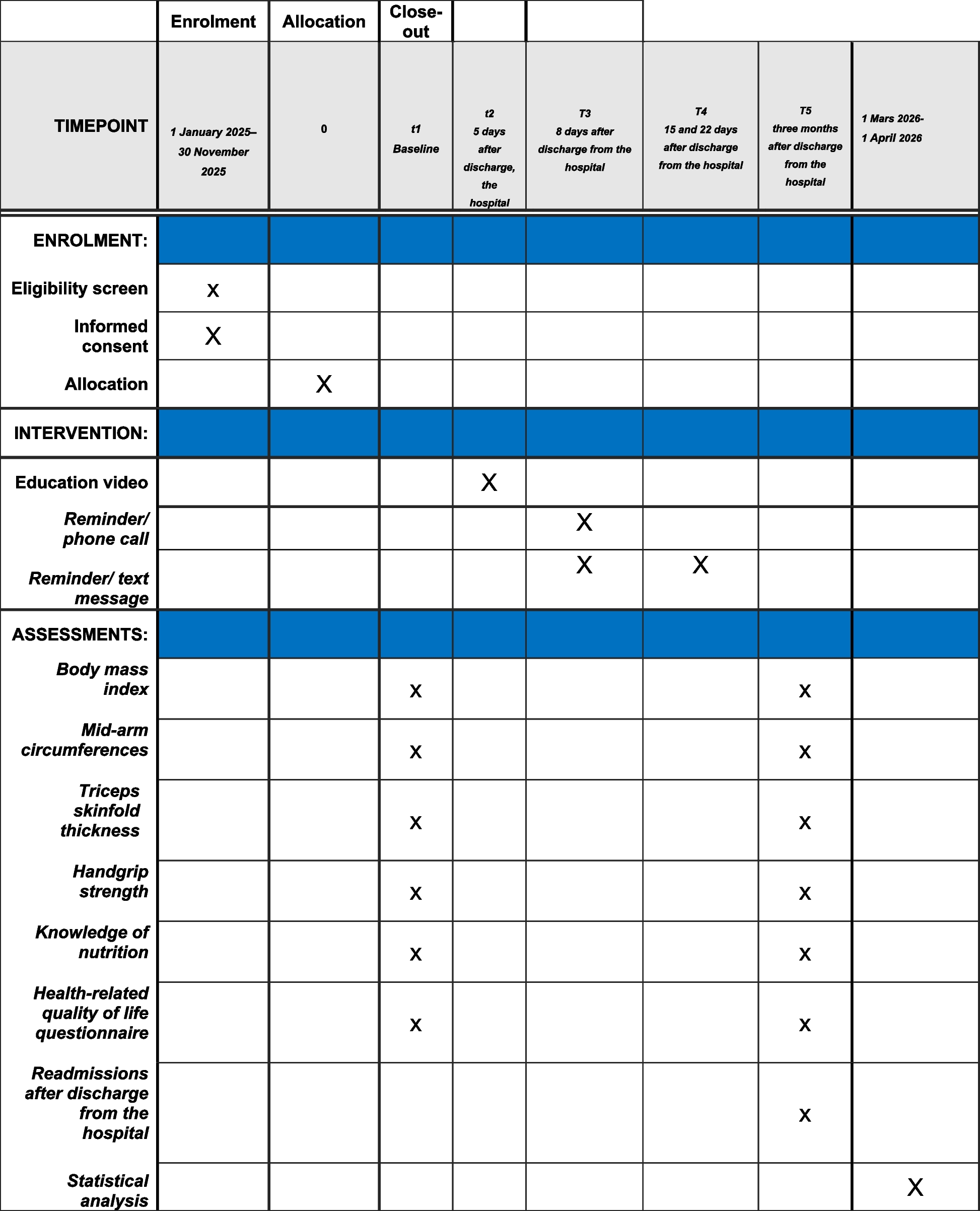
Participant timeline
Preoperative visit
ICU admission
Day 1 till ICU discharge
ICU discharge
Beyond ICU discharge
Informed consenta
X
X
Randomization and start of study intervention
X
Measurement of blood glucose and titration of insulin according to the allocated intervention
X
X
Blood and urine sampleb
X
X
Clinical assessment of muscle weakness and ICU functional statusc
X
Electrophysiological and morphological assessment of muscle weakness, and needle biopsyd
X
Discontinue study interventione
X
Assessment of long-term survival and functional status
X
aDepending on whether the admission is elective or urgent, and whether one is able to contact the patient before ICU admission, informed consent is obtained prior or shortly after ICU admission (deferred informed consent)
bBlood and urine samples are taken as per routine practice. Routine daily measurements include routine clinical chemistry, hematology, and markers of inflammation. Other routine measurements are only determined on selected days, as per local practice. In selected centers, extra biological samples are taken upon admission (blood samples) and thereafter daily (blood and urine samples) for research purposes. In selected centers, samples of pericardial and pleural fluid are collected upon admission and thereafter daily, in patients admitted after cardiac surgery, as described in the protocol
cThese tests are performed on selected days in ICU, as described in the protocol
dThis is performed on selected days in ICU (in selected centers, depending on additional funding), as described in the protocol
eThe study intervention is discontinued upon ICU discharge or earlier, in case the patient is able to resume oral feeding or when the central venous catheter is removed and not replaced
Sample size
To detect a reduction in ICU dependency by 1 day with at least 80% power (two-tailed) and 95% certainty, and assuming a baseline mean ICU stay of 9 days and a standard deviation of 15, 2782 patients in each group need to be included (total 5564). For safety reasons, we also want to exclude any clinically relevant harmful impact on hospital mortality (safety endpoint). To detect an adversely increased hospital mortality from 8.5 to 10% with 80% power and 95% certainty, 4612 patients per group need to be included (total 9224). Hence, we plan to include 9230 patients.
The baseline mean ICU stay and its standard deviation are based on our previous multicenter RCT studying the impact of early parenteral nutrition, which was performed in the same or comparable centers in the same healthcare system [17]. As the baseline ICU stay may have changed over time and may differ per center, and as it is difficult to predict the relative contribution of each center, an interim analysis was planned after inclusion of 25% of the study population (n=2308), to allow adjustment of sample size if needed, based on the observed data in the control group. At that time, the independent data monitoring committee indicated that there was no need to adjust the sample size.
Recruitment
All patients admitted to one of the participating ICUs are screened for eligibility upon ICU admission (within 2 h).

留言 (0)