
記住我


Six- to eight-week-old male C57BL/6 mice (15–20 g) were maintained in a standard environment at 22 ± 1 °C on a 12-h light/dark cycle. After adaptive feeding for 1 week, mice were subjected to the modeling. The experimental protocol for UC-CRC model establishment was illustrated in Fig. 1A. In brief, mice were injected with 10 mg/kg AOM (A5486, Sigma, China) intraperitoneally. One week later, mice were given 3% DSS (MP Biomedicals, China) in drinking water for consecutive 7 days, followed by normal drinking water for consecutive 14 days. The DSS-normal drinking water cycle was repeated for 3 times. To determine the role of Pou3f1 in UC-CRC development, the adeno-associated virus 9 vectors carrying short hairpin RNA targeting Pou3f1 (AAV-shPou3f1) and negative control (AAV-shNC) were constructed. The AAV particles were administrated to UC-CRC mice by coloclysis 2 weeks prior to AOM treatment (Fig. 2A).
Fig. 1
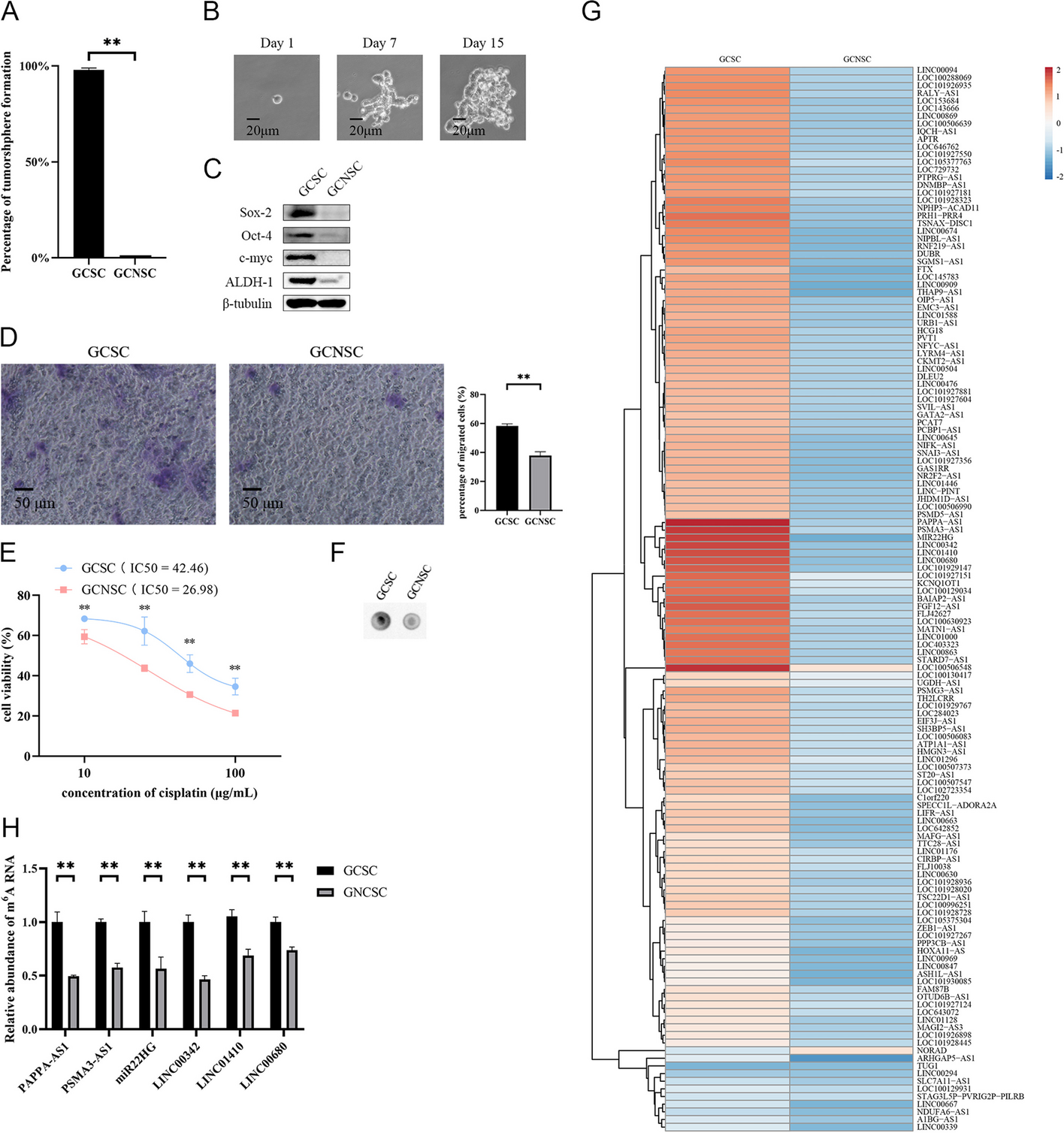
Pou3f1 physically interacted with Nfatc3, and it was upregulated in colons of UC-CRC mice. (A) The schematic diagram for animal model protocol. (B-C) The colon length in mice was recorded. (D) Representative images of HE-stained colon sections. (E) Nfatc3 mRNA levels in colons were determined using qPCR. (F) Western blot analysis for Nfatc3 protein levels in colons and quantitative results. (G-H) ChIP-PCR and ChIP-qPCR were performed to determine the interaction between Nfatc3 and Pou3f1 in colons. M: Marker. (I) ChIP-qPCR assay showed that Nfatc3 bound to the Src promoter, rather than the Cdx2 promoter in colon tissues. Src was used as a positive control. Cdx2 was used as a negative control. (J) The relative mRNA levels of Pou3f1 were detected by qPCR analysis. Data were from n = 6 mice per group. Values were mean ± SD. **, p < 0.01
Fig. 2
Pou3f1 was identified as a direct transcriptional target of Nfatc3. (A) The schematic of the potential motifs and binding sites of Nfatc3 in the Pou3f1 promoter region. (B-C) ChIP-PCR and ChIP-qPCR were performed to demonstrate the direct interaction between Nfatc3 and Pou3f1 in RAW264.7 cells. M: Marker. (D) ChIP-qPCR assay demonstrated that Nfatc3 bound to the Src promoter, rather than the Cdx2 promoter in RAW264.7 cells. Src was used as a positive control. Cdx2 was used as a negative control. (E) The relative luciferase activity was measured by luciferase reporter assay in RAW264.7 cells. (F) The mRNA levels of Pou3f1 in RAW264.7 cells and BMDMs were measured by qPCR. (G) Western blot analysis and quantification results for Pou3f1 protein levels in RAW264.7 cells and BMDMs. n = 3. Values were mean ± SD. *, p < 0.05; ** p < 0.01
Several indicators for animal health were monitored weekly, including body weight, drinking water/food consumption, stool, diarrhea, and rectal bleeding. The disease activity index was determined as previously described [24].
Cell treatmentRAW264.7 cells (ZQ0098) were purchased from ZhongQiaoXinZhou (China) and cultured in Dulbecco’s modified eagle medium (DMEM; G4510, Servicebio, China) containing 10% FBS with 5% CO2 at 37 °C. Mouse bone marrow-derived macrophages (BMDMs; iCell-0060a, iCell Bioscience Inc, China) were incubated in a special complete medium (CM-M141, Procell, China) with 5% CO2 at 37 °C.
For gene overexpression or knockdown in vitro, the adenovirus vectors expressing Nfatc3/control (Ad-Nfatc3/Ad-Vector) and short hairpin RNA targeting Pou3f1/control (Ad-shPou3f1/Ad-shNC) were prepared to infect RAW264.7 cells or BMDMs. The infection was conducted for 48 h via Lipofectamine 3000 (L3000-008, Invitrogen, USA). After the 48-h infection, RAW264.7 cells were treated with 100 ng/ml lipopolysaccharide (LPS; L8880, Solarbio, China) for 3 h to induce an acute inflammatory response.
Chromatin immunoprecipitation (ChIP) assayChIP assay was performed using the Tissue ChIP Kit (WLA122, Wanleibio, China) or the Cell ChIP Kit (WLA106a, Wanleibio) following the manufacturers’ protocols. Colon tissues were cut into 1–3 mm pieces, crosslinked with 1% formaldehyde and placed in a glass homogenizer for homogenizing. RAW264.7 cells were crosslinked with 1% formaldehyde. Chromatin was immunoprecipitated using Nfatc3 antibody (700 µg/mL; 18222-1-AP, Proteintech, China) or normal rabbit IgG antibody (0.5 µg/µL). The precipitated DNAs were subjected to PCR or qPCR reaction. The primer sequences (5’-3’) targeting Pou3f1 were: forward, AGCCAAATGATGGACAGA; reverse, GGTATGAGATAGAGGGAGTG.
Luciferase reporter assayTo assess the effect of Nfatc3 on Pou3f1 transcriptional activity, the fragments of the wild type or mutant Pou3f1 promoter region were inserted into pGL3-Basic luciferase reporter vectors. The Pou3f1 luciferase reporters were co-transfected with Nfatc3 overexpression plasmids (Nfatc3-OE) into RAW264.7 cells using Lipofectamine 3000. Forty-eight hours later, firefly and renilla luciferase activities were measured using the Luciferase Reporter Gene Assay Kit (KGAF040, KeyGEN BioTECH, China).
Measurement of ROS levelsThe ROS Assay Kit (S0033, Beyotime, China) was utilized to measure intracellular ROS contents in vitro. Cells were washed in PBS twice, and incubated with DCFH-DA solution at 37 °C. After resuspending in PBS, the fluorescence intensity was determined by flow cytometry (NovoCyte, AceaBio, USA).
To detect ROS production in vivo, colon tissues were cut into small pieces and crushed with a homogenizer in PBS to prepare single-cell suspensions. After incubation with DCFH-DA solution (E004, JianCheng, China) at 37 °C, the single-cell suspensions were examined with a multimode Reader (Synergy H1, Biotek, USA). The fluorescent intensity of ROS was measured at the excitation wavelength of 490 nm and the emission wavelength of 540 nm.
Enzyme-linked immunosorbent assay (ELISA)For ELISA analysis, colon segments were mechanically crushed in saline and homogenate supernatants were harvested after centrifugation. The protein concentration of colon supernatants was quantified with a BCA Protein Assay Kit (PC0020, Solarbio). The amount of IL-1β (EK201B, Multi Sciences, China), monocyte chemotactic protein-1 (MCP-1; EK287, Multi Sciences), and prostaglandin E2 (PGE2; EK8103, Multi Sciences) in colon homogenate was determined by ELISA.
Determination of MPO activityColons were homogenized in the MPO buffer and the homogenate suspensions were extracted. The MPO activity was determined by an MPO Activity Assay Kit (A044, JianCheng).
Histological stainingColons were harvested, fixed in paraformaldehyde and embedded in paraffin. After deparaffinization, the 5-µm sections were stained with hematoxylin (H8070, Solarbio) and eosin (A600190, Sangon, China) (HE) for histological analysis as previously described [25]. For immunohistological staining, the sections were incubated with COX-2 antibody (1:50 dilution; A1253, ABclonal, China), and then incubated with HRP-conjugated Goat anti-Rabbit secondary antibody (1:500 dilution; 31,460, ThermoFisher Scientific, USA). For immunofluorescent staining, primary antibodies against Proliferating Cell Nuclear Antigen (PCNA; 1:50 dilution; A0264, ABclonal) and F4/80 (1:50 dilution; sc-377,009, Santa Cruz, USA) were applied. Sections were then incubated with secondary antibodies, including FITC-labeled Goat anti-Rabbit IgG antibody (1:200 dilution; A0562, Beyotime) and Cy3-labeled Goat anti-Mouse IgG antibody (1:50 dilution; A0521, Beyotime). TUNEL staining was performed to determine cell death in colons using the In Situ Cell Death Detection Kit (11,684,795,910, Roche, Switzerland) following the manufacturer’s instructions. Cell nuclei were counterstained using DAPI (D106471, Aladdin, China). Images were taken using the Olympus microscope (BX53, Olympus, Japan). The number of positive cells within 3 random ×400 fields/each Sect. (2 sections per mouse) was assessed by manual counting.
Quantitative real-time PCR (qPCR)Total RNA from the ground colon tissues in nitrogen and cultured cells was extracted using the TRIpure Reagent lysis buffer (RP1001, BioTeke, China), followed by cDNAs reverse transcription with the BeyoRT II M-MLV reverse transcriptase (D7160L, Beyotime). qPCR was performed with the SYBR Green reagent (SY1020, Solarbio) on the Exicycler 96 system (Bioneer, Korea). The primer sequences (5’-3’) were shown: Nfatc3 forward GGTAAAGAGCAGCACATA, Nfatc3 reverse TTGACTAGAGGCAGGATT; MCP-1 forward GCCTGCTGTTCACAGTTGCC, MCP-1 reverse CTGGACCCATTCCTTCTTGG; TNF-α forward CAGGCGGTGCCTATGTCTCA, TNF-α reverse GCTCCTCCACTTGGTGGTTT; IL-6 forward ATGGCAATTCTGATTGTATG, IL-6 reverse GACTCTGGCTTTGTCTTTCT; IL-1β forward CTCAACTGTGAAATGCCACC, IL-1β reverse GAGTGATACTGCCTGCCTGA; Pou3f1 forward CGTGTTCTCGCAGACCACCATC, Pou3f1 reverse CGCACCACCTCCTTCTCCAGTT; GAPDH forward TGTTCCTACCCCCAATGTGTCCGTC, GAPDH reverse CTGGTCCTCAGTGTAGCCCAAGATG. The relative gene expression was calculated with the 2−ΔΔCt method and normalized to GAPDH.
Western blotWestern blot analysis was performed as previously reported [26]. Total protein was prepared with the RIPA lysis reagent (R0010, Solarbio). After quantification with the BCA Protein Assay Kit, protein samples were separated on the SDS-PAGE gel and transferred onto PVDF membranes (IPVH00010, Millipore, USA). Membranes were incubated with primary antibodies, including Nfatc3 antibody (1:1000 dilution; 18222-1-AP, Proteintech), Pou3f1 antibody (1:1000 dilution; A19330, ABclonal), iNOS antibody (1:1000 dilution; A0312, Abclonal), COX-2 antibody (1:1000 dilution; A1253, Abclonal) and GAPDH antibody (1:10000 dilution; 60004-1-Ig, Proteintech). The Goat anti-Rabbit IgG/HRP antibody (1:3000 dilution; SE134, Solarbio) and Goat anti-Mouse IgG/HRP antibody (1:3000 dilution; SE131, Solarbio) were used as secondary antibodies. The protein signals were visualized using the ECL Western Blot Substrate (PE0010, Solarbio).
Statistical analysisAll data were expressed as mean ± SD and analyzed using GraphPad Prism Software. Unpaired t test or one-way ANOVA followed by Bonferroni’s multiple comparisons test was used to determine the statistical difference. Survival analysis was performed using Kaplan-Meier with the Logrank test. p < 0.05 was identified as a significant difference.
留言 (0)