Cell lines and culture condition
Human TNBC cell lines MDA-MB-231 (ATCC, Manassas, VI, USA) and HCC1395 (ATCC), murine TNBC cell line 4T1 (ATCC) and human THP-1 monocytes (ATCC) were maintained in RPMI-1640 medium (Life Technologies, Grand Island, NY, USA) with 10% fetal bovine serum (FBS) (Life Technologies), 2 mM l-glutamine (HyClone, South Logan, UT, USA), 100 units/ml penicillin (HyClone) and 100 μg/ml streptomycin (HyClone) in a 37 °C incubator with 5% CO2.
Primary antibodies for Western blotting
Ab against p21 was purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). Antibodies (Abs) against Arginase-1, cleaved PARP (Asp214), EGFR, iNOS, IL-10, PARP, p38 MAPK, phospho-AMPK (Thr172), phospho-EGFR (Tyr1068), PD-L1, phospho-Histone H2AX (Ser139) (γ-H2AX), phospho-p38 MAPK (Thr180/Tyr182), phospho-PKM2 (Tyr105), PKM2 and Rad51 were obtained from Cell Signaling (Danvers, MA, USA). Abs against β-actin, AMPK and IL-6 were purchased from GeneTex, Inc. (Irvine, CA, USA). Abs against GluT1, HK2, PFKL, RPIA and MCT4 were obtained from Elabscience (Houston, TX, USA). All the above Abs were used for Western blotting. SDS–PAGE and immunoblotting were conducted as described previously [32].
Oligo-Fucoidan preparation
Oligo-Fucoidan (also known as Low-Molecular-Weight Fucoidan LMF) was purified from Laminaria japonica seaweed sample using similar approaches as previously described [29, 30]. Briefly, 100 g of dried seaweed power were suspended in 5 liter of distilled water and boiled at 100 °C for 30 min, centrifuged at 10,000×g for 20 min, and the supernatant was incubated with 4 M CaCl2 for 1 h to separate alginic acid by centrifugation at 10,000×g for 20 min. All the supernatants were dialyzed in deionized water for 48 h in a molecular weight (MW) cut-off of 10 kDa dialysis tube. The crude Fucoidan extracts were precipitated by ethanol at a ratio of 1:3 (V/V) and then fractionated through anion‐exchange chromatography using DEAE‐Sephadex A‐25 (Sigma–Aldrich) equilibrated with 20 mM Tris–HCl, pH8.0 and eluted with 1.5 M NaCl as described by Hwang et al. [30], and the fraction 3 of eluates were hydrolyzed with the crude glycolytic enzyme isolated from Bacillus subtilis [30], into Oligo-Fucoidan with an average MW of 1.2 kDa (~ 90.1%). Oligo-Fucoidan was composed of sulfate (35.4% ± 1.3% (w/w)) and neutral monosaccharides, including fucose (38.71 ± 0.41%), glucose (6.09 ± 0.52%), galactose (19.19 ± 0.31%), myo-inositol (2.95 ± 0.54%), mannose (28.78 ± 0.71%), xylose (3.46 ± 0.34%) and rhamnose (0.82 ± 0.30%). The ration of monosaccharide contents were calculated from the following equation: mole of monosaccharide/mole of (fucose + glucose + galactose + myo-inositol + mannose + xylose + rhamnose) × 100%. The purified Oligo-Fucoidan was dissolved in phosphate-buffered saline (PBS), stirred for 30 min at room temperature and sterilized using filtration before performing experiments.
Mammosphere formation
4T1 and MDA-MB-231 cells (5 × 104) were cultured in 6-well ultralow attachment plates (Corning) (Corning, NY, USA) in serum-free DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MI, USA) supplemented with 1% l-glutamine, 1% penicillin/streptomycin, 2% B27 (Invitrogen, Carlsbad, CA, USA), 20 ng/ml EGF (Sigma–Aldrich, St. Louis, MO, USA) and 20 ng/ml FGFb (PeproTech, Rehovot, Israel) and treated with 50 μM olaparib (Selleckchem, Houston, TX, USA) and/or 400 μg/ml Oligo-Fucoidan for 5 or 16 days, respectively. The number of mammospheres (≥ 50 μm) was counted using ImageJ software (http://rsb.info.nih.gov/ij/index.html). Photographs were taken with a Nikon DIAPHOT300 inverted microscope at magnifications of 100 × (for 4T1 cells) and 200 × (for MDA-MB-231 cells). All experiments were performed in triplicate, and mammospheres were quantified in 8–10 randomly selected fields.
Mammosphere cells (1 × 105) were harvested and further stained with Alexa Fluor® 647-conjugated anti-human CD24 Ab (BD Pharmingen, San Jose, CA, USA), FITC-conjugated anti-human CD44 Ab (BD Pharmingen) or BB515-conjugated anti-human CD326 Ab (EpCAM) (BD Pharmingen) for 1 h on ice, rinsed with PBS, resuspended in 500 μl of PBS and analyzed by the BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). CD44-FITC and CD326-BB515 were excited at 488 nm, and the emissions were measured by FL1 PMT (515–545 nm bandpass filter). CD24-Alexa 647 was excited at 633 nm, and the emission was measured by FL-4 PMT (653–669 nm bandpass filter). CellQuest software (BD Biosciences) and FlowJo software (BD Biosciences) were used to determine the subpopulations of CD44(+)/CD24(−) and EpCAM(+) cells, respectively.
Colony assay of cell viability
TNBC cells were plated on 6-well plates (5 × 103 cells) and treated 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan for 14 days. Cell colonies were fixed with 4% paraformaldehyde in PBS for 30 min at 4 °C, washed three time with PBS, stained with 0.2% crystal violet for 2 h, photographed with an optical microscope and quantified by ImageJ software.
WST-1 cell viability assay
MDA-MB-231 cells were treated with olaparib (0 ~ 50 μM) or 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan for 24 h and incubated with WST-1 (4[3-(4-iodophenyl)-2(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) assay solution (Sigma–Aldrich) for 1 h. The tetrazolium salt was cleaved to form formazan by cellular mitochondrial dehydrogenase, the formazan dye was quantitated with a scanning multiwell spectrophotometer. The measured absorbance directly correlates to the number of viable cells.
Cell cycle analysis
MDA-MB-231 cells were treated with 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan for 48 h. The cells were fixed with 70% ethanol at − 20 °C for 1 h and resuspended in PBS containing 0.1% (v/v) Triton X-100 (Sigma–Aldrich), 5 μg/ml DNase-free RNase A (Sigma–Aldrich) and 10 μg/ml propidium iodide (PI) (Thermo Fisher Scientific) in the dark for 30 min. PI fluorescence was excited at 543 nm, and the emissions at 615 nm were measured by the FL2 PMT channel and quantified with the BD FACSCalibur flow cytometer.
Quantitative real-time polymerase chain reaction (qRT–PCR)
Total RNA was extracted using TRIzol™ reagent (Invitrogen). The RNA samples were quantified using a NanoDrop ND1000 spectrophotometer (Thermo Fisher Scientific). An aliquot of total RNA (1 μg) was reversely transcribed using a Maxima First Strand Synthesis kit (Thermo Fisher Scientific) at 25 °C for 10 min, 50 °C for 15 min and 85 °C for 5 min before chill on ice for 10 min. qRT–PCR was conducted using SYBR Green Master Mix (Roche Diagnostics, Basel, Switzerland) and analyzed by an ABI ViiA Thermal Cycler (Applied Biosystems, Foster City, CA, USA). qRT–PCR was performed at 95 °C for 15 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. β-actin level was used as an internal control. Relative mRNA level was calculated by the following formula: ΔΔCT = ΔCt test sample-ΔCt control sample. Fold changes in gene expression were calculated using the comparative 2−ΔΔCT method.
The primers were synthesized by PURIGO Biotechnology (Taipei, Taiwan). The primer sequences of amplicons for human ALDH1A1 (forward: 5′-CACGCCAGACCTACCTGTCC-3′; reverse: 5′-GCAGAGCTCCTCAGTTG-3′), CD44 (forward: 5′-TTACAGCCTCAGCAGAGCAC-3′; reverse: 5′-TGACCTAAGACGGAGGGAGG-3′), CD24 (forward: 5′-CTCACAGAACAAAGCAAGGGC-3′; reverse: 5′-GCCTAGCGCGAACCCTCC-3′), EpCAM (forward: 5′-TGCTGGAATTGTTGTGCTGG-3′; reverse: 5′-AGATGTCTTCGTCCCACG-3′), Nanog (forward: 5′-TGGGAAGAAGCTAAAGAGCCAG-3′; reverse: 5′-GGATGCTTCAAAGCAAGGCA-3′), Sox2 (forward: 5′-CATGAAGGAGCACCCGGATT-3′; reverse: 5′-TTAATGTGCGCGTAACTGTG-3′), Snail (forward: 5′-GCGAGCTGCAGGACTCTAAT-3′; reverse: 5′-GGACAGAGTCCCAGATGAGC-3′), F4/80 (forward: 5′-CAATGAGTGCCTCACCAGCA-3′; reverse: 5′-TGGGCAAGCTCTTGGATCTG-3′), CD80 (forward: 5′-GCAGGGAACATCACCATCCA-3′; reverse: 5′-TCACGTGGATAACACCTGAACA-3′), CD86 (forward: 5′-GCTTTGCTTCTCTGCTGCTG-3′; reverse: 5′-GGCAGGTCTGCAGTCTCATT-3′), CD163 (forward: 5′-CCGGGAGATGAATTCTTGCCT-3′; reverse: 5′-AGACACAGAAATTAGTTCAGCAGCA-3′), CD206 (forward: 5′-CTGAATTGTACTGGTCTGTCCT-3′; reverse: 5′-GCTTAGATGTGGTGCTGTGG-3′), TGF-β (forward: 5′-TTGACTTCCGCAAGGACCTC-3′; reverse: 5′-CTCCAAATGTAGGGGCAGGG-3′) and β-actin (forward: 5′-CACCAGGGCGTGATGGTGGG-3′; reverse: 5′-GATGCCTCTCTTGCTCTGG GC-3′) were designed according to the NCBI Probe database.
Analysis of the macrophage polarity
Human THP-1 monocytes (1 × 105) were treated with 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan for 48 h. Polarized macrophages were stained with FITC-conjugated anti-CD80 Ab and Alexa Fluor 647-conjugated anti-CD163 Ab (BD Pharmingen) were diluted (1:50) in staining buffer (1% FBS in PBS) for 1 h on ice. Fluorescence intensity was quantified by the BD FACSCalibur flow cytometer in the FL1 and FL4 channels at excitation/emission wavelengths of 488 nm and 633 nm, respectively. The results were quantified using FlowJo software.
THP-1 monocytes were treated with 100 ng/ml phorbol myristate acetate (PMA) (GeneTex, Inc.) for 72 h to induce M0 macrophage differentiation. M0 macrophages were stained with Alexa Fluor 546-conjugated anti-F4/80 Ab (BD Pharmingen) excited by a 488-nm laser line, and the emissions were measured in the FL2 channel (564–606 nm) with a 585/42 BP filter by the BD FACSCalibur flow cytometer.
MDA-MB-231 cells (4 × 105) were pretreated with 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan for 48 h, the treatments were rinsed off, conditioned medium (CM) was collected, and the CM was incubated with M0 macrophages for 48 h. The phenotypes of CD80(+) M1 and CD163(+) M2 macrophages were measured by flow cytometry as described above.
Trajectories of cancer cell migration
MDA-MB-231 cells (1 × 105) were treated with 50 μM olaparib and/or 400 μg/ml Oligo-Fucoidan in RPMI medium containing 0.5% serum for 48 h. Cell mobility was analyzed with a Leica AF6000LX microscope (Leica Microsystems, Wetzlar, Germany) using a 20 × objective. Images were acquired for 18–24 h at 10-min intervals. Cell movements were tracked with Metamorph (Molecular Devices, San Jose, CA, USA) to quantify the total migration distances. Cell trajectories emanating from the initial position were plotted using the DiPer macro [33].
Glucose uptake assay
A glucose uptake cell-based assay kit (Cayman Chemical, Ann Arbor, MI, USA) was used to analyze cellular glucose uptake. Briefly, cells (5 × 104/well) were seeded in 96-well plates and incubated overnight in 100 μl of glucose-free medium, reacted with 200 μg/ml 2-NBDG (2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-d-glucose) for 1 h in glucose-free medium and then centrifuged for 5 min at 400×g at room temperature to remove the supernatant. Two hundred microliters of the assay buffer was added to each well, the plate was centrifuged for 5 min at 400×g at room temperature, the supernatant was aspirated, and 100 μl of assay buffer was added to each well. 2-NBDG fluorescence was quantified using the infinite M200 PRO (TECAN, Männedorf, Switzerland) with excitation/emission wavelengths of 485/525 nm.
Lactate production assay
An l-Lactic acid (LA) colorimetric assay kit (ACE biolabs, Taoyuan, Taiwan) was used to measure LA levels. Briefly, 4T1 or MDA-MB-231 cells (4 × 105) were cultured in medium containing 10% FBS, treated with the indicated agents for 24 h and incubated in medium containing 3% FBS for another 24 h. The cells were collected and resuspended in 100 μL of PBS, sonicated on ice and collected the supernatants after centrifugation at 1500×g for 10 min at 4 °C. Protein concentrations were determined by Coomassie Plus Protein Assay Reagent (Thermo Fisher Scientific). The samples were mixed with the enzyme working solution and chromogenic agent and incubated at 37 °C for 10 min. The stop solution was added, and the absorbance was measured at 530 nm by a microplate reader (TECAN). LA levels were calculated by the following formula: LA (mmol/gm protein) = (ΔOD1 sample/ΔOD2 standard) × 3 (the concentration of standard, mmol/l) × dilution factor of sample/concentration of protein in sample (gm of protein/l).
4T1 TNBC progression, metastasis and recurrence
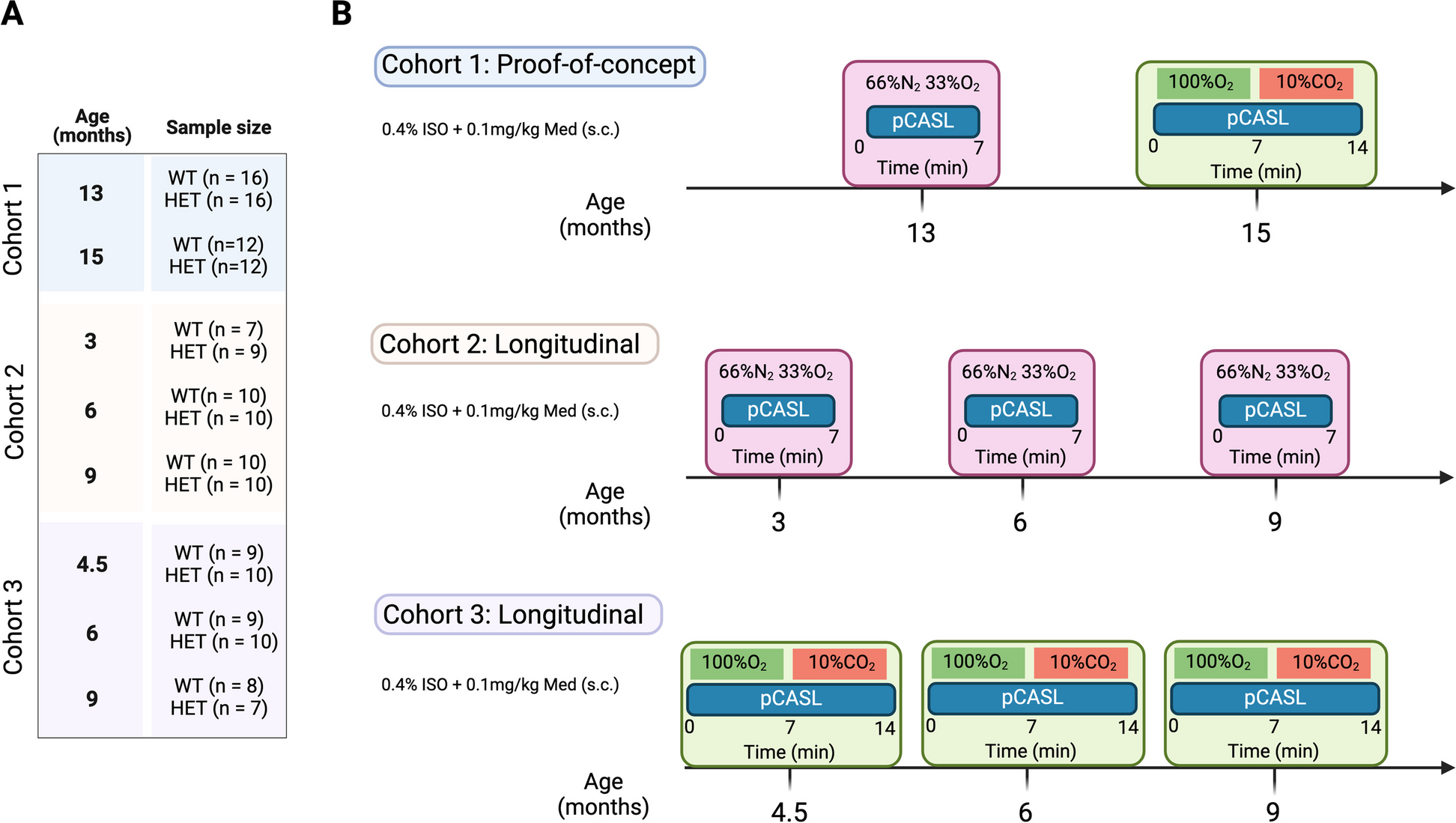
Six-week-old female BALB/c mice were obtained from the National Laboratory Animal Center (Taipei, Taiwan) and approved by the Animal Use Protocol (NHRI-IACUC-108026-A). 4T1 cancer cells (1 × 104) were implanted in the 4th mammary fat pad. Tumor volume was measured weekly as previously described [34].
To evaluate postsurgical therapeutic outcomes, primary tumors were surgically removed when the tumor volumes reached approximately 150 mm3 (week 2). Xenograft mice were intraperitoneally injected with olaparib (50 mg/kg) twice per week for 2 weeks or were administered Oligo-Fucoidan (150 mg/kg) by oral feeding twice per week for 5 weeks. PBS was used as a control treatment. At week 7, tumor relapse, metastasis and mouse mortality rates were evaluated. Recurrent and metastatic tumors were processed for further analysis as previously described [34].
Immunohistochemical (IHC) analysis
IHC analysis of tumors was performed using a DAKO EnVision detection kit (Agilent Technologies, Santa Clara, CA, USA). Anti-CD163 Ab (E-AB-70306) (Elabscience Biotechnology Inc, Houston, TX, USA) (1:600) was added and incubated with the specimens for 30 min, after which the samples were washed with PBS for 15 min and incubated with a labeled polymer-HRP-conjugated secondary Ab (DAKO EnVision System) for 10 min. The Ab-reactive samples were visualized using 3,3′-diaminobenzidine substrate (DAKO EnVision System), counterstained with Mayer’s hematoxylin (Sigma–Aldrich), dehydrated with ethanol (70%, 3 min; 80%, 3 min; 95%, 3 min; and 100%, 5 min), cleared with xylene (5 min), and cover-slipped (Automat-star 24 × 50 mm) with Histokitt solution mounting medium (Glaswarenfabrik Karl Hecht GmbH & CO KG., Germany). IHC images were observed under a Nikon Optiphot-2 upright microscope with a 40 × objective lens and analyzed by the automatic digital slide scanner system Pannoramic MIDI II (3DHISTECH, Ltd., Budapest, Hungary). ImageJ Fiji software (version 1.2) (NIH, Bethesda, MD, USA) was used to conduct color deconvolution to adjust the image threshold and analyze the signal intensity as described previously [35]. The results were evaluated in at least 6 randomly chosen fields.
Flow cytometric analysis of splenic lymphocytes
The olaparib and/or Oligo-Fucoidan-treated 4T1 cell xenograft mice were sacrificed at week 7. The spleen tissue was harvested and rinsed by PBS, gently minced in ice-cold RPMI-1640 (Life Technologies) containing 2% FBS (Life Technologies), and then mashed using the plunger of a 1 ml syringe. The cell suspension was passed through a 70-μm-nylon cell strainer to remove clumps, lysed with red blood cell lysis buffer (155 mM NH4Cl, 12 mM NaHCO3 and 0.1 mM EDTA), and fixed with 4% (v/v) paraformaldehyde (Alfa Aesar, Ward Hill, MA, USA). The cell suspension (1 × 106) was then resuspended with anti-mouse CD16/CD32 Ab (1:2000) in 0.1% bovine serum albumin (Sigma–Aldrich) in ice-cold PBS to block nonspecific binding and labeled with cocktails of biotin-conjugated mouse anti-mouse NK-1.1, rat anti-mouse CD19 Ab, rat anti-mouse CD4 Ab, and rat anti-mouse CD8a Ab at a 1:100 dilution. The labeled cells were purified by positive selection using Streptavidin Particles Plus. Purified lymphocytes were stained with PE mouse anti-mouse NK-1.1 Ab (1:25), APC rat anti-mouse CD19 Ab (1:50), PerCP-Cy5.5 rat anti-mouse CD4 Ab (1:50) and BB515 rat anti-mouse CD8a Ab (1:100). The isotype control Abs of PE hamster IgG2 κ, BB515 rat IgG2b κ, APC rat IgG2a κ and PerCP-Cy5.5 rat IgG2a κ were used as negative controls. All the biotinylated and conjugated Abs used for flow cytometry analysis of the lymphocytes were purchased from BD Biosciences. Analysis was performed using the BD FACSCalibur flow cytometer and FlowJo v10. BB515 rat anti-mouse CD8a Ab, PE mouse anti-mouse NK-1.1 Ab and PerCP-Cy5.5 rat anti-mouse CD4 Ab were excited by a 488-nm laser line, and the emissions were measured in the FL1 channel (515–545 nm) with a 530/30 BP filter, FL2 channel (564–606 nm) with a 585/42 bandpass (BP) filter and FL3 channel with a 670 nm long-pass (LP) filter, respectively. APC rat anti-mouse CD19 Ab was excited by a 635-nm laser line, and the emission was measured in the FL4 channel (653–669 nm) with a 661/16 BP filter.
Flow cytometric analysis of tumor-infiltrating Tregs
Recurrent breast tumor tissues were minced and enzymatically digested with a cocktail containing 1 mg/ml collagenase D (Sigma–Aldrich), 0.25 mg/ml DNase I (Sigma–Aldrich), and 0.25% (v/v) trypsin–EDTA solution (HyClone, Logan, UT, USA) in serum-free RPMI-1640 medium (Life Technologies). Similar with lymphocyte preparation, a single-cell suspension (1 × 106) was blocked with anti-mouse CD16/CD32 (1: 2000) (Sigma–Aldrich) and then positively selected by anti-mouse CD4 magnetic particles and then stained with PerCP-Cy5.5 rat anti-mouse CD4 Ab (1:50), PE rat anti-mouse CD25 Ab (1:25) and APC rat anti-mouse CD127 Ab (1:50). The isotype controls Abs (PE hamster IgG2 κ, APC rat IgG2a κ and PerCP-Cy5.5 rat IgG2a κ) were used as negative controls. Analysis was performed using the BD FACSCalibur flow cytometer and FlowJo v10. Tregs were defined as CD25(+)/CD127(−) subsets gated from CD4(+) populations. PE rat anti-mouse CD25 Ab and PerCP-Cy5.5 rat anti-mouse CD4 Ab were excited by a 488-nm laser line, and the emissions were measured in the FL2 channel (564–606 nm) with a 585/42 BP filter and the FL3 channel with a 670 nm LP filter, respectively. APC rat anti-mouse CD127 Ab was excited by a 635-nm laser line, and the emission was measured in the FL4 channel (653–669 nm) with a 661/16 BP filter. All the conjugated Abs were purchased from BD Biosciences.
Statistics
The data are presented as the means ± S.E.M. One-way ANOVA with Newman–Keuls test or Duncan’s test was used to analyze the data. *p < 0.05, **p < 0.01, ***p < 0.001.

留言 (0)