
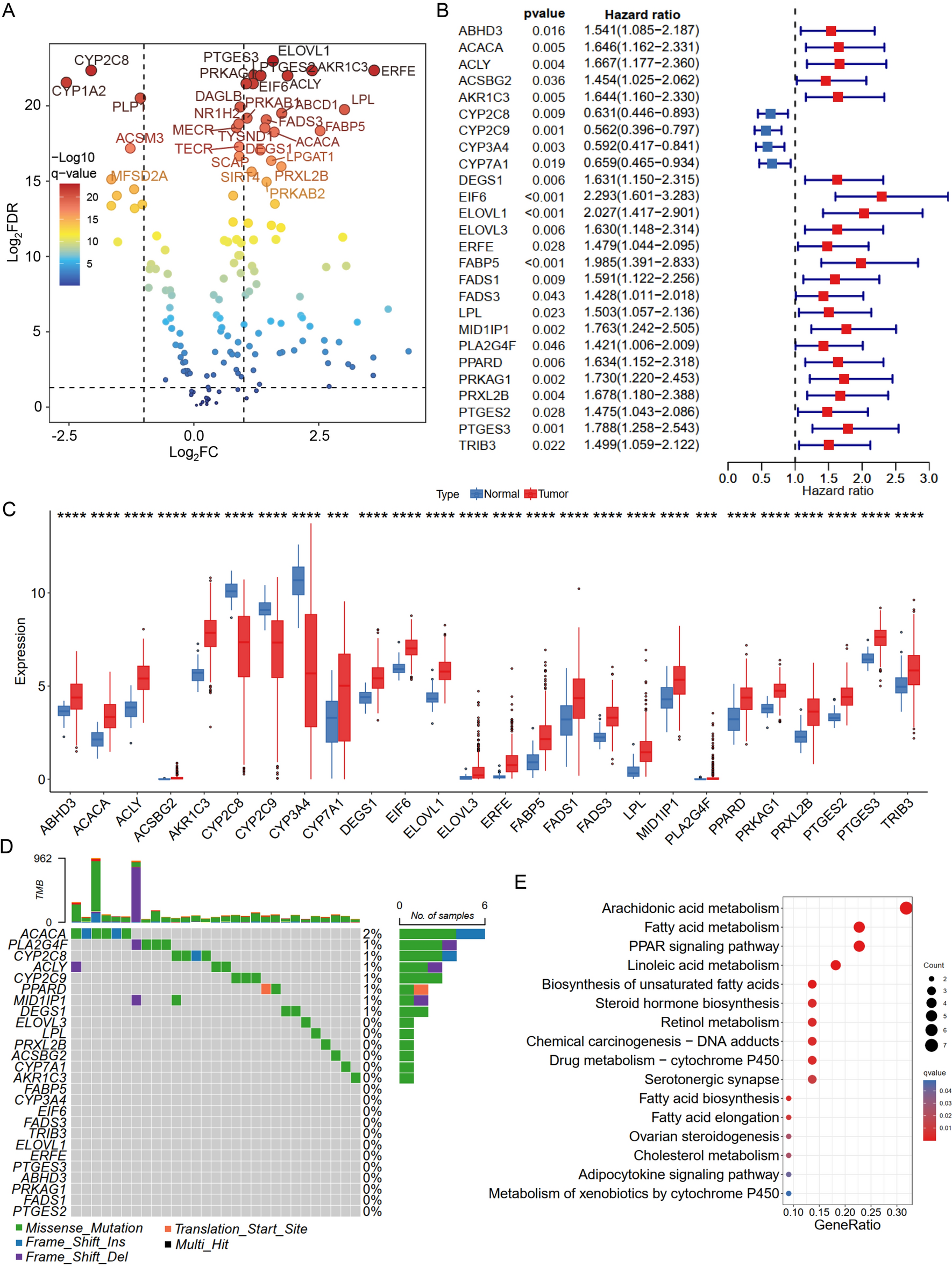
記住我
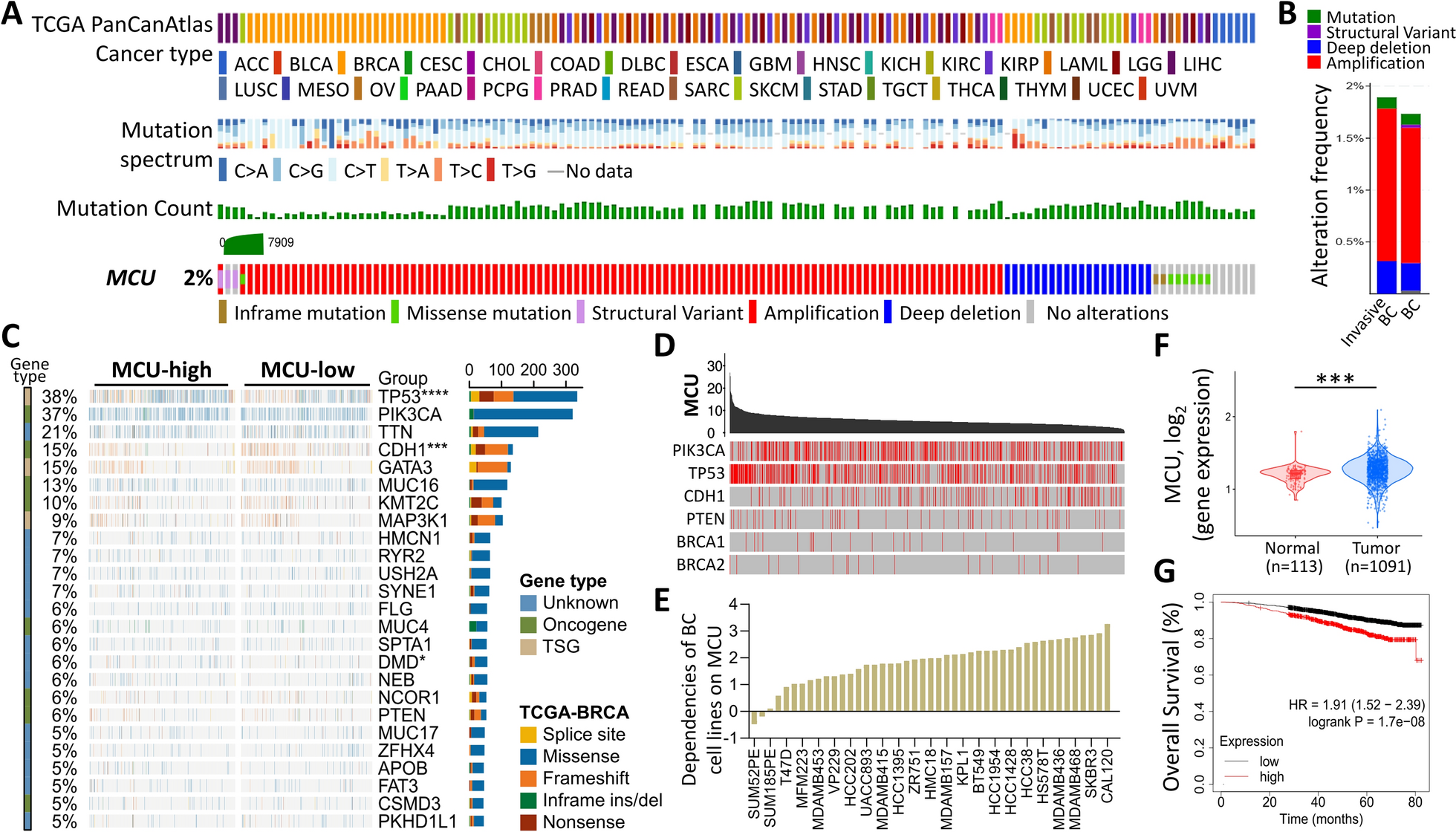
Long non-coding RNAs (lncRNAs) have been largely investigated for their contribution in human disorders, particularly cancer [1]. These transcripts have sizes more than 200 nucleotides, do not possess considerable open reading frames and regulate gene expression through diverse epigenetic mechanisms. They participate in transcriptional and post-transcriptional regulation via interacting with DNA, RNA or proteins [2]. Moreover, they are involved in the regulation of mRNA splicing and can serve as precursors for microRNAs (miRNAs) [3]. Thus, lncRNAs regulate gene expression at almost all levels.
Maternally expressed 8 (MEG8), alternatively named as RNA Imprinted and Accumulated in Nucleus (Rian), is an example of which is expressed in the nucleus [4, 5]. In human, MEG8 gene resides in a cluster of imprinted genes on chromosome 14q32.3. The encoded transcript is has a preferential expression from the maternal allele in skeletal muscle, and seems to be regulated in a coordinate manner with other imprinted genes in this genomic area (https://www.ncbi.nlm.nih.gov/gene/79104). It is highly expressed in adrenal, placenta and brain [6]. This small nucleolar RNA host gene has 52 known splice variants (https://asia.ensembl.org/Homo_sapiens/Gene/Splice?db=core;g=ENSG00000225746;r=14:100894770-101038859).
Recent studies have shown contribution of MEG8 in different disorders ranging from neoplastic ones to diabetic nephropathy, atherosclerosis, ischemic stroke, trophoblast dysfunction and abortion, Henoch-Schonlein purpura and osteoarthritis. In the current review, we summarize the role of this lncRNA in mentioned disorders, based on the evidence obtained from in vitro, in vivo and human studies.
In vitro studiesLiu et al. have investigated function of MEG8 in lung cancer. For this purpose, they have transfected lung epithelial BEAS-2B cells with MEG8 overexpressing vector. Moreover, they have transfected lung cancer A549 and H1299 cells with MEG8 or miR-107 overexpressing vectors as well as knockdown plasmids. Up-regulation of MEG8 has increased proliferation, migration and invasion of lung epithelial cells. On the other hand, MEG8 knockdown or miR-107 up-regulation has blocked cell progression of lung cancer cells. Their functional studies have confirmed competitive binding of MEG8 and CDK6 with miR-107 and their function in regulation of progression of lung cancer. In addition, MEG8 knockdown or miR-107 overexpression could suppress Rb and E2F3 phosphorylation. Taken together, MEG8 could enhance progression of lung cancer through regulation of miR-107/CDK6 axis and activation of Rb/E2F3 pathway [7]. Another study in lung cancer has shown up-regulation of MEG8, parallel with down-regulation of miR-15a-5p and miR-15b-5p in cancer cell lines. MEG8 silencing has suppressed proliferation, migration, and invasion of lung cancer cells through targeting miR-15a-5p/miR-15b-5p [8].
Terashima et al. have shown induction of expression of MEG8 in the course of TGF-β-mediated epithelial-mesenchymal transition (EMT) in both lung and pancreatic cancer cells. Up-regulation of MEG8 could suppress expression of miR-34a and miR-203, leading to over-expression of SNAI1 and SNAI2 transcription factors and subsequent repression of cadherin 1/E-cadherin. Mechanistically, MEG8 interacts with EZH2 protein and increases recruitment of EZH2 to the regulatory sections of miR-34a and miR-203. EZH2 enhances histone H3 methylation in these regions and suppress transcription of these miRNA genes. Concurrent expression of MEG8 and MEG3 can increase EMT-associated alterations in cell morphology and increase motility of cells in the absence of TGF-β. Taken together, MEG8 participates in the induction of EMT through epigenetic mechanisms [9].
The effects of MEG8 silencing have also been investigated in human hemangioma endothelial cells. Notably, MEG8 silencing has suppressed proliferation of these cells and increased their apoptosis through modulation of the effects of miR-203 on JAG1 and Notch expressions [10]. Figure 1 summarizes the effect of MEG8 in the pathogenesis of lung cancer and hamangioma.
Fig. 1
Oncogenic roles of MEG8 in lung cancer and hemangioma
Expression of MEG8 has been found to be elevated in hepatocellular carcinoma (HCC) cells. MEG8 silencing has significantly suppressed the proliferative and invasive abilities of these cells. Furthermore, MEG8 has been shown to sponge miR-367-3p to increase 14-3-3ζ levels, suppress degradation of TGFβR1, and promote TGF-β signaling [11].
Moreover, MEG8 has been shown to participate in the progression of bone-invasive pituitary adenoma through sponging miR-454-3p and increasing TNF-α expression [12]. Similarly, expression of MEG8 has been found to be elevated in Wilms tumor cells, parallel with up-regulation of CRK and down-regulation of miR-23a-3p. MEG8 silencing or miR-23a-3p up-regulation has blocked viability, migration potential and invasive properties of these cells. Mechanistically, MEG8 binds with miR-23a-3p to release CRK from inhibitory effects of this miRNA. Taken together, MEG8 regulates pathogenesis of Wilms tumor through miR-23a-3p/CRK axis [13]. Figure 2 shows the oncogenic roles of MEG8 in hepatocellular carcinoma, bone invasive pituitary adenoma and Wilms tumor.
Fig. 2
Oncogenic roles of MEG8 in hepatocellular carcinoma, bone invasive pituitary adenoma and Wilms tumor
Exposure of podocyte cells with high-glucose conditions has led to over-expression of MEG8 and miR-770-5p. In fact, up-regulation of MEG8 increases miR-770-5p levels through decreasing methylation of the miR-770-5p gene. Up-regulation of MEG8 and miR-770-5p can increase cell apoptosis under high-glucose conditions. Taken together, MEG8 can increase miR-770-5p levels via epigenetic mechanism to induce diabetic nephropathy through enhancing cell apoptosis [14].
MEG8 can also contribute in the pathogenesis of other non-neoplastic conditions. For instance, it participate in the pathoetiology of atherosclerosis through regulation of proliferation, migration and apoptosis of vascular smooth muscle cells via affecting expression of PPARα [15]. This lncRNA can attenuate cerebral ischemia following ischemic stroke via influencing miR-130a-5p/VEGFA axis [16].
Over-expression of MEG8 in trophoblast cells has reduced proliferation and invasion of these cells, while its silencing has exerted the opposite effects. This imprinted lncRNA participates in the modulation of function of trophoblast cells during early stages [17].
MEG8 can also contribute in the pathogenesis of Henoch Schonlein purpura through sponging miR-181a-5p, influencing levels of SHP2 expression and increasing M1 macrophage polarization [18]. Finally, this lncRNA can regulate proliferation and apoptosis of chondrocytes, thus participating in the pathogenesis of osteoarthritis [19].
MEG8 has also been shown to contribute to the pathoetiology of cardiovascular diseases through epigenetic mechanisms. In vitro studies have demonstrated that MEG8 knock-down impairs angiogenic sprouting and reduces proliferation of HUVEC cells. RNA sequencing experiments have shown up-regulation of the inhibitor of angiogenesis TFPI2 after MEG8 silencing. From a mechanistical point of view, MEG8 silencing can lead to a decrease in H3K27me3 marks at the TFPI2 promoter [20]. Table 1 shows summary of in vitro studies about the role of MEG8 in human disorders.
Table 1 Summary of in vitro studies about the role of MEG8 in human disorders (∆: knock-down or deletion, VSMC: vascular smooth muscle cell, OGD: oxygen-glucose deprivation)
留言 (0)