Cat eye syndrome is a chromosomal disorder associated to a supernumerary marker chromosome due to simultaneous duplication and inversion of chromosome 22, resulting in its partial tetrasomy or trisomy. The anomaly usually appears sporadically and may occur during gametogenesis, mainly oogenesis, or as an early post-zygotic event [3].
The proximal region of the long arm of chromosome 22 (22q11.2) is critical for chromosomal rearrangements, due to its increased content of highly homologous repetitive regions, known as low copy repeats (LCRs), more prone to recombination events during meiosis. 22q11 region, besides CES, is implicated in other congenital malformation syndromes, like DiGeorge (DGS) and Derivative 22 syndrome (der 22). Considerable overlap may exist between CES and other syndromes with anal, auricular, heart and urogenital anomalies, like Townes–Brocks syndrome, which must be included in the differential diagnosis [5]. Fourteen genes have been identified to date in the CES critical region, and located in the proximal 22q11 chromosome. Among them, CECR1 (Cat Eye Syndrome chromosome region candidate 1 or Adenosine Deaminase 2, MIM 607575) and CECR2 (Cat Eye Syndrome chromosome region candidate 2, MIM 607576), have been considered as candidates for involvement in the duplication phenotype (heart and facial defects, and brain and eye development anomalies respectively [6]).
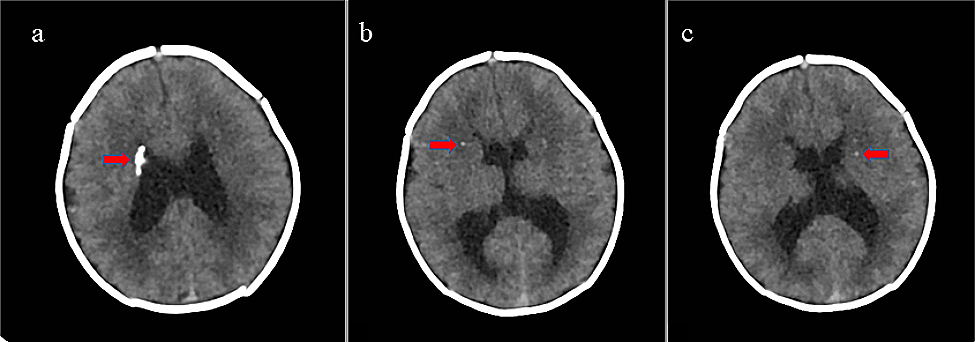
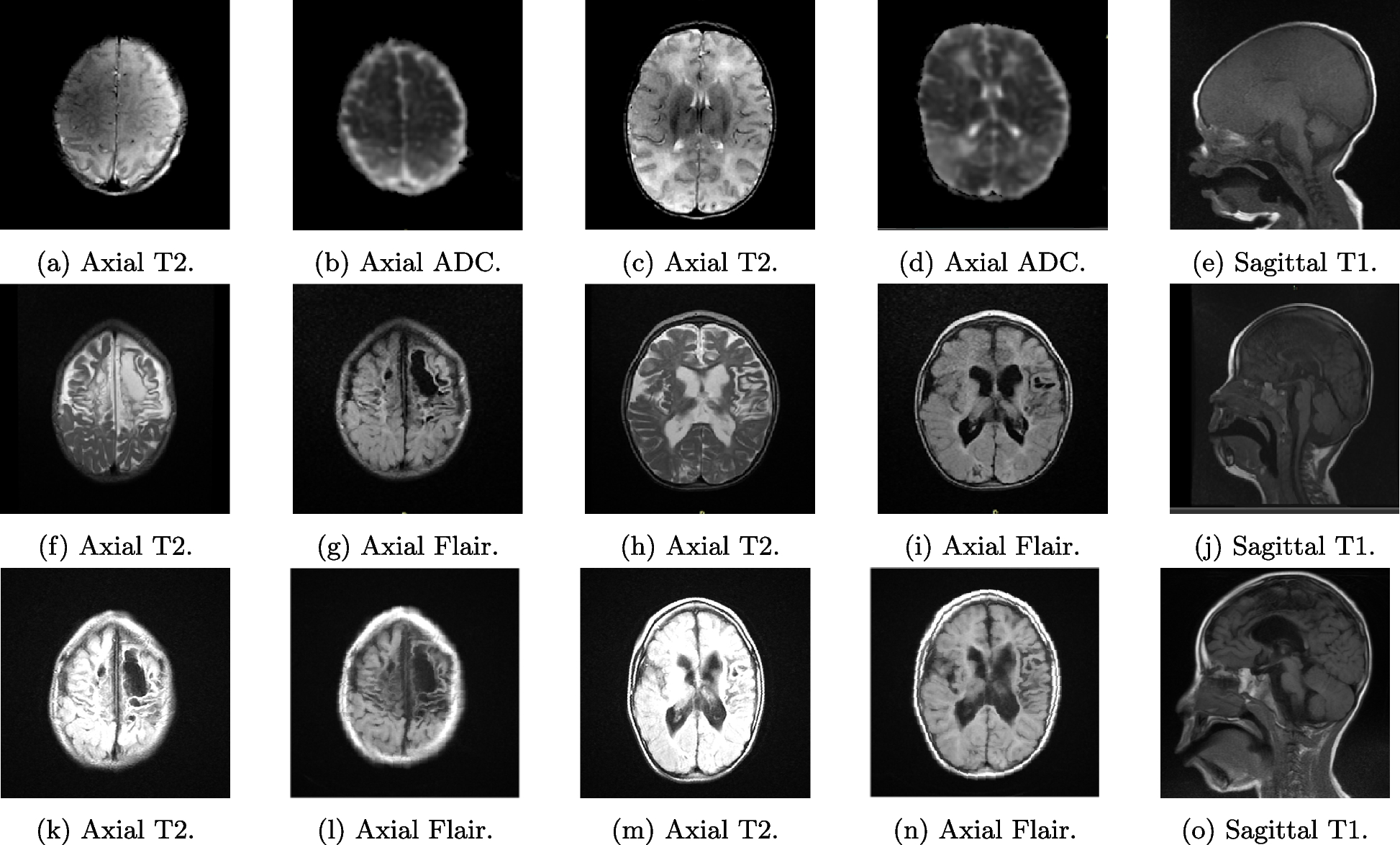
Our patient showed a classical CES phenotype, which included iris and choroid colobomas, anal malformation and ear anomalies. Such classic triad is found in about 40% of affected subjects. In addition, our newborn manifested persistent hypoglycemia and subsequent cholestasis, associated to multiple hormone deficiencies. Transitional hypoglycemia of healthy newborns must be distinguished from that persisting or occurring for the first time beyond the first 3 days of life. Indeed, persistent hypoglycemia most often results from congenital or genetic defect in the regulation of insulin secretion, cortisol and/or growth hormone deficiencies, or inborn errors of metabolism affecting glucose, glycogen, or fatty acids [7]. Measuring bicarbonate, lactic acid, beta-hydroxybutyrate, free fatty acids (FFA), insulin and carnitine levels during hypoglycemia (blood glucose < 50 mg/dL) is useful in differentiating among metabolic causes, hypoglycemic hyperinsulinism and fatty acids oxidation disorders [8]. In our case, glucagon test did not elicit any increase in insulinemic response, ruling out hyperinsulinism. Moreover, hormone deficiencies allowed us to identify the correct pathophysiological mechanism underlying hypoglycemia, suggesting thus hypopituitarism. The latter was then confirmed by brain MRI, which showed aplasia of the anterior pituitary gland, abnormal stalk, and ectopic neurohypophysis (triad known as “Pituitary Stalk Interruption Syndrome”, PSIS) [9].
Hypopituitarism is the second cause of persistent hypoglycemia in neonates. Conversely, neonatal cholestasis as a sign of hypopituitarism is rare [10]. Cortisol increases bile flow, and its deficiency may cause abnormalities in the synthesis and transport of bile acids, leading in some cases to cholestasis. Our patient, indeed, had very high serum bile acids levels, and the biochemical evidence of cholestatic hepatitis with increased levels of direct and total bilirubin, GGT and transaminases (Table 2) was observed on the thirteenth day of life, according to literature data [11]. Once cortisol replacement treatment is started, cholestasis disappears in around 10 weeks [9]. In our case, in fact, the resolution of cholestasis with hydrocortisone replacement therapy occurred within an overlapping time frame, moreover suggesting a causal relationship between them both.
22q11.2 rearrangements may influence the development of midline structures, including the pituitary gland [12], and other midline brain anomalies (absent/hypoplastic corpus callosum and septum pellucidum, schizencephaly, eterotopia) may be associated to PSIS, as in our newborn who also showed corpus callosum hypoplasia. Associated growth hormone deficiency and/or hypothalamic–pituitary axis malformations are, however, poorly documented [13], and the few cases of the literature reporting on CES patients showing hormonal defects and/or brain MRI anomalies are summarized in Table 3.
Table 3 Endocrine profile and/or brain (hypothalamic-pituitary region) MRI anomalies of present patient compared to those reported in literatureThe pathogenic mechanisms underlying hypopituitarism and/or other endocrine defects associated to CES may be searched in the genes included within the chromosomal rearrangements of the affected subjects. Specifically in our proposita, the partial chromosome 22q tetrasomy involves around 30 genes [17], and most of them are highly expressed in the brain and endocrine glands (mainly gonads, in addition to adrenals and thyroid). Particular attention should be paid to those which may alter embryonic development, the formation of median axes of the brain and of the whole midline, and which then may be related to hypothalamic-pituitary anomalies and hypopituitarism. Among these, ADA2 regulates cell proliferation and differentiation and could be linked, besides the phenotypic features of the syndrome, also with its endocrine abnormalities. TUBA8 (tubulin alpha 8, MIM 605742) shows major transcription in heart, testicle and brain. Its mutations are associated with polymicrogyria and hypoplasia of the optic nerve, and then a correlation with defects of other nearby structures as the pituitary stalk may be supposed. Finally, DGCR6 (DiGeorge syndrome critical region gene 6, MIM 601279) and DGCR5 (DiGeorge syndrome critical region gene 5, MIM 618040, partially involved in our patient), which are associated with DiGeorge syndrome and participate in gonadal and germ cell development, are highly expressed in heart, testis, thyroid and brain [18]. Therefore, their possible causal relationship with hypopituitarism and/or other endocrine defects may be considered. Other mechanisms cannot be ruled out (many uncharacterized or not coding genes are comprised in the critical region, with potential positional or regulatory effects), and, ultimately, although a common genetic basis between pituitary anomalies and CES seems plausible, to date it remains to be elucidated.
Our report underlines how genetic diseases may be burdened by additional hidden malformations and/or dysfunctions. Although poorly documented, CES patients may have pituitary gland anomalies with endocrine defects/dysfunctions, including cortisol, thyroxine, GH and/or ACTH deficits, as occurred in the present patient. In addition, they need extensive endocrine screening including GH levels evaluation, also in light of the possibility of GH replacement treatment, which may improve child growth and final height [12].
In cases of newborns or infants with persistent hypoglycemia and cholestasis, neonatologists and pediatricians must be aware of the possible occurrence of hypopituitarism. When hypoglycemia is detected, early intervention is essential to minimize the risk of poor neurologic outcomes and developmental delay [19, 20]. We strongly recommend glycemic and hormone screening of CES patients, to prevent potential life-threatening conditions. A multidisciplinary (auxological/endocrinological, neurodevelopmental, surgical, ophthalmological, audiological, cardiological, orthopedic) management and longitudinal follow-up [21,22,23,24,25,26,27] must be guaranteed to affected subjects. The latter should be oriented to a prompt recognizing of complications and/or associated anomalies [28,29,30,31,32,33,34], allowing practitioners to thus lower mortality rates and short- and long-term adverse outcomes [35,36,37].





留言 (0)