
記住我
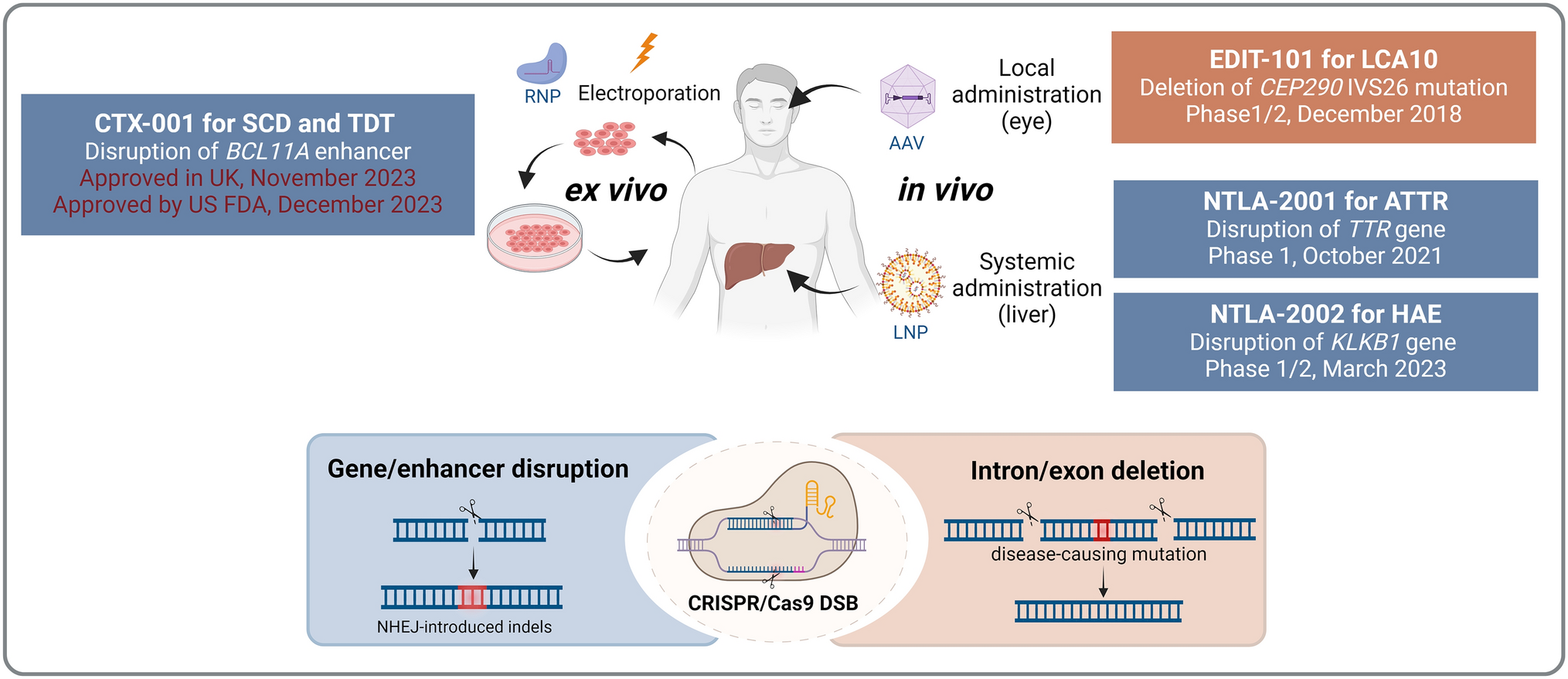

Although there are approximately 20 proteins causing systemic amyloidosis, in 95% of patients the disease is caused either by LC or TTR. Efforts to develop anti-amyloid antibodies have been directed toward these two proteins, in addition to pan-amyloid approaches.
The amyloid heart involvement is the determinant of survival in AL and ATTRwt amyloidoses and the main determinant in patients with ATTRv amyloidosis with mixed heart and nerve phenotypes. Removing amyloid deposits from the heart is the main target of passive immunotherapy. One of the critical factors in developing effective passive immunotherapy is the choice of target (epitope). The design of the anti-amyloid antibodies was different for each antibody tested in the clinic (Table 1). All therapeutic monoclonal antibodies used in trials in systemic amyloidosis, either anti-plasma cells or anti-fibrils, are immunoglobulin (Ig) G1 due to the capability of this subclass to activate complement, mediate complement-dependent cytotoxicity, and recruit effector cells for antibody-dependent cellular cytotoxicity against human target cells. Furthermore, this subclass binds to the human neonatal Fc receptor (FcRn) that protects IgG1 molecules from degradation and thereby extends their serum half-life. Additionally, human IgG1 antibodies are stable, form fewer aggregates, and have favorable biotechnological characteristics that facilitate their production and cost containment [26].
Table 1 Characteristics of monoclonal antibodies targeting amyloid deposits used in interventional clinical trialsTargeting Common Constituents of Amyloid DepositsDezamizumab and MiridesapPepys and collaborators pursued the brilliant project to develop an antibody capable of recognizing and promoting the resorption of amyloid deposits regardless of the protein precursor, exploiting the presence of the normal plasma protein serum amyloid P component (SAP) as a common constituent of all amyloid deposits [27]. The therapy included the depletion of SAP from plasma by the drug (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanol]pyrrolidine-2-carboxylic acid (CPHPC, miridesap). This drug is a competitive inhibitor of SAP binding to amyloid fibrils and also crosslinks and dimerizes SAP molecules, leading to their very rapid clearance by the liver, and thus produces a marked depletion of circulating human SAP [28]. The safety, tolerability, pharmacokinetics, and pharmacodynamics of miridesap alone have been investigated in both healthy volunteers and patients with systemic amyloidosis (NCT01323985, NCT02953808, NCT01406314), with a lack of treatment-related adverse events or safety signals and a dose-dependent depletion of circulating SAP up to > 95% of normal levels [29, 30].
CPHPC leaves some SAP in amyloid deposits that can be specifically targeted by therapeutic IgG anti-SAP antibodies. The activation of the classic complement pathway by the monoclonal IgG1 anti-SAP-antibodies leads to the opsonization of amyloid deposit and attracts a massive number of macrophages that fuse into multinucleated giant cells that reabsorb amyloid deposit, as observed in an animal model of amyloid A type amyloidosis [31].
The humanized IgG1 anti-SAP antibody (dezamizumab) in combination with miridesap has been tested in an open-label, single dose-escalation, phase I trial (NCT01777243) (Table 2), which aimed to evaluate the safety and pharmacokinetic profile of this combination therapy [32]. Patients with different types of systemic amyloidosis and with evidence of splenic and/or hepatic amyloid involvement were eligible to enter this study, while subjects with cardiac amyloid involvement were not included for safety reasons. The first part of the trial was a single-center, open-label, single dose-escalation study testing intravenous administration of dezamizumab (from 0.1 to 30 mg/kg) combined with miridesap. Results on 15 treated patients (8 AL, 4 AFib, 2 AA, 1 AApoAI) were reported (Table 3) [32]. No serious adverse events emerged during this study. Most patients receiving > 200 mg of dezamizumab experienced infusion-related symptoms, including, but not limited to, headache, a sensation of warmth, flush, changes in heart rate and blood pressure, nausea, abdominal discomfort, diarrhea, or loose stools. These manifestations were self-limiting and did not persist longer than 12 h after antibody infusion. A more than 200 mg dose of dezamizumab was also accompanied by a systemic response, with a brief spike in serum interleukin (IL)-6 and IL-8 at 2 h, a peak in C-reactive protein and serum amyloid A protein, and a transient, approximately twofold increase in peripheral-blood neutrophil count at 24 h in most cases. Pharmacokinetic measurements demonstrated a rapid depletion of circulating SAP upon miridesap treatment, as expected, as well as rapid disappearance of dezamizumab from the circulation and depletion of serum C3, consistent with prompt sequestration of the therapeutic antibody by amyloid deposits (Table 1) and classic complement pathway activation, as reported in the above-mentioned AA mouse study [31]. Even though the study was not designed to assess efficacy, the amyloid load was monitored and at 42–45 days after the single administration of dezamizumab, there was evidence of a decrease in liver stiffness at elastography, a reduced extracellular volume at MRI, and reduced hepatic/renal/lymph nodal amyloid load by 123I-SAP-scintigraphy in a subset of cases [32].
Table 2 Clinical trial designTable 3 Clinical trial outcomesThe second part of the study consisted of an adaptatively extended antibody dosing, evaluating up to three treatments of dezamizumab (up to 2000 mg) at intervals of at least 2 months, to investigate dose-response effects (Table 2). Based on the acceptable safety profile of the first part of the study, patients with cardiac amyloidosis were eligible for the second part, even though NYHA III/IV classes, decompensated cardiac failure, a recent history of syncope/presyncope, or NT-proBNP >1800 ng/L were among the exclusion criteria. The 15 patients evaluated in the first part of the trial went on to continue treatment and 8 additional patients were included, for a total of 23 patients (12 AL, 5 AFib, 3 ATTR, 2 AA, 1 AapoAI) evaluated (Table 3) [33]. Of note, all included AL patients were in complete response or very good partial response after chemotherapy at the beginning of the study (although three of these patients experienced a hematologic relapse during the study) and the underlying inflammatory conditions of the two AA patients were in complete remission. A total of 48 treatments were performed (1 in 8 patients, 2 in 5 patients, and 3 in 10 patients). Overall, there were three serious adverse events, including hypotension, tachycardia, and transient elevation of serum creatinine in one patient, an erythema multiform-like rash in a second patient, and a spontaneously resolving episode of fast atrial fibrillation in a third patient with a history of episodic atrial fibrillation. Dose-related infusion reactions and mild to moderate urticaria or macular rashes were seen in most recipients of dezamizumab ≥ 600 mg. Two skin biopsies were performed, one showing dermal leukocytoclastic vasculitis (LCV) and the other demonstrating only non-specific changes. Pharmacokinetic studies confirmed the observations from the first part of the trial, with dezamizumab concentration rapidly decreasing, particularly in patients with heavy amyloid load and C3 depletion. A decrease in liver stiffness, reduced extracellular volume, and reduced hepatic, renal, splenic, or adrenal amyloid load by 123I-SAP-scintigraphy were documented in a subset of cases, particularly after infusion with the highest doses of dezamizumab. Amyloid clearance from the liver occurred faster and was more conspicuous compared with other organs. Among the six patients with cardiac amyloidosis (three AL and three ATTR), in four cases a transient, up to fivefold increase in serum NT-proBNP concentration was noted, which lasted for about 1 week and was taken to suggest the engagement of the target mechanism in the heart. However, no systematic change in left ventricular mass by cardiac MRI, or change in cardiac uptake at 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) scintigraphy (for the three ATTR cases) was documented [33].
Based on these results, a larger study with serial anti-SAP dosing at regular and closer intervals was designed to extend safety and establish the efficacy of miridesap/dezamizumab combination therapy in cardiac amyloidosis (NCT03044353) (Table 2). This was a single-arm, open-label, phase II study evaluating monthly repeat anti-SAP treatment in systemic amyloidosis participants with cardiac dysfunction caused by cardiac amyloidosis. The study was planned to include three groups of patients [34]. Group 1 consisted of patients with cardiac ATTR (either ATTRwt or ATTRv); group 2 included patients with cardiac AL amyloidosis having already achieved at least a very good partial response to previous chemotherapy and not requiring further anti-plasma cell therapy at study initiation; and group 3 was deemed to include newly diagnosed, cardiac AL patients having attained a free LC complete response after three cycles of chemotherapy, with at least the first cycle consisting of a combination of cyclophosphamide, bortezomib, and dexamethasone. Patients with NT-proBNP levels exceeding 8500 ng/L who had a very poor median survival of 4 months [19] were excluded from the trial. The experimental treatment consisted of an initial infusion of miridesap on days − 2 and − 1 (which could eventually be repeated once daily up to day 3) to achieve a serum concentration of SAP below 3 mg/L, followed by dezamizumab infusion (600–1200 mg, split into two doses at day 1 and day 3), accompanied by up to three-times-daily subcutaneous injections of miridesap from day 1 to day 11 (Table 3). Six dezamizumab treatments per patient were planned, with 1-month intervals between each session. Six patients from group 1 with cardiac ATTR amyloidosis received between three and six treatment sessions (total dezamizumab dose between 3000 and 6000 mg). The presence of anti-dezamizumab antibodies could be demonstrated in all patients receiving four or more treatment sessions at the 8-week follow-up and persisted at 6-month follow-up visits. This high rate of antidrug antibody formation is higher than expected for a humanized antibody [35]. The isotype of these anti-dezamizumab antibodies and their potential impact on dezamizumab bioavailability, pharmacokinetic/pharmacodynamic properties, efficacy, and safety was not reported [35,36,37].
Treatment-related adverse events occurred in all but one of the seven enrolled patients, the most frequent consisting of an urticarial and/or maculopapular rash, with histopathological evidence of LCV in both cases where a skin biopsy was performed. Notably, in the first AL patient enrolled in group 2, following initial dezamizumab dosing (300 mg), a treatment-related serious adverse event occurred, consisting of a presumptive abdominal large-vessel vasculitis (aortitis) (Table 3) [34]. It is of interest that SAP is also a normal component of several basement membranes, including the glomerular, alveolar, and some vascular basement membranes [38,39,40]. The potential binding of the mAb to SAP in vessels is supposed to be the basis of dermal LCV, manifesting as a rash. There is the potential for overlap between dermatological manifestations of LCV and an event of large-vessel vasculitis [34]. Regarding efficacy data, no consistent change in left ventricular mass, left ventricle wall thickness or extracellular volume at cardiac MRI could be documented. Furthermore, there was no consistent pattern of change in cardiac functional measures or NT-proBNP levels during the study period [34].
A concurrent immune positron emission tomography (PET) study on patients with cardiac amyloidosis (NCT03417830) showed only moderate to low cardiac uptake of 89Zr-dezamizumab on the first two cardiac ATTR amyloidosis patients enrolled [34]. In addition to impaired cardiac dezamizumab microvascular perfusion, this may also be a result of the continuous structure of the endothelium in cardiac tissue, in contrast to the fenestrated endothelium found in the liver and spleen, the organs in which the most striking treatment effects had been previously reported [32]. The concentration of dezamizumab in cardiac tissue may have been insufficient to trigger macrophage and giant cell recruitment and amyloid clearance. This observation raises the general critical question of the therapeutic antibodies' access to cardiac amyloid deposits. Occurrence of the treatment-related serious adverse event in conjunction with the lack of cardiac signal of efficacy led to a reconsideration of the benefit–risk assessment for the treatment, and both clinical trials and the development of this treatment for cardiac amyloidosis were terminated [34].
AT-01, AT-02, AT-03Similar pan-amyloid approaches, although not based on targeting amyloid deposits with antibodies, have been developed by Wall and collaborators. A radiolabeled polybasic peptide (also known as AT-01) that binds via electrostatic interactions to electronegative glycosaminoglycans and protein fibrils, two common components of amyloid deposits, is being used for imaging systemic amyloidosis in AL and ATTR amyloidosis in a phase II study (NCT05235269) [41]. This peptide was fused with the Fc portion of IgG1 immunoglobulin (AT-02) to promote the phagocytic reabsorption of amyloid deposits [42]. The preclinical characterization of a novel fusion protein of SAP with IgG1 Fc portion (AT-03), with pan-amyloid binding and removal properties, showed that a single intravenous dose of AT-03 resulted in a significant reduction of mouse splenic AA amyloid within 14 days post-injection [43].
Targeting the Amyloid Deposits in AL AmyloidosisBirtamimabThe murine monoclonal antibody 2A4, whose humanized form is birtamimab, binds an epitope derived from a cleavage site of serum amyloid protein A containing a -Glu-Asp- amino acid pairing [44]. The antibody also binds LC amyloid fibrils, both κ, and λ. Plasmon resonance analyses using synthetic AL fibrils as a substrate revealed that 2A4 bound with a KD of 10 nM. Radiolabeled 2A4 specifically localized with human AL amyloid extracts implanted in mice (amyloidomas) as shown by single-photon emission computed tomography (SPECT) imaging. Treatment with 2A4 accelerates regression of AL κ amyloidomas in mice, compared with animals that received a control antibody [45].
A subsequent study reported that 2A4 bound both soluble and insoluble LC aggregates extracted from patient tissue, and induced macrophage engagement and phagocytic clearance of AL amyloid deposits in vitro [46].
Birtamimab (NEOD001) was tested in a first-in-human, uncontrolled, open-label, multicenter, phase I/II study on AL amyloidosis patients with persistent amyloid organ dysfunction despite having achieved partial hematologic response or better to at least one previous line of anti-plasma cell therapy (NCT01707264) (Table 2) [47]. This trial aimed first at establishing the maximum tolerated dose or phase II recommended dose of birtamimab when infused as a single agent in patients with AL amyloidosis, and then at evaluating birtamimab safety, preliminary efficacy, and pharmacokinetics in the same clinical setting. Patients with NT-proBNP levels above 5000 ng/L, a life expectancy < 3 months, the presence of a co-existing, symptomatic multiple myeloma, and patients requiring plasma cell-directed therapy were excluded from the trial. The dose-escalation phase of the study had a 3 + 3 design, with seven dose cohorts covering the range of 0.5–24 mg/kg, until the achievement of the maximum tolerated dose (defined as the highest dose level at which no more than one of six patients experienced dose-limiting toxicity during the first month of therapy). Birtamimab was administered every 28 days, with a treatment duration of up to 1 year. The interim results on the 27 patients enrolled in the dose-escalation phase of the study were initially reported [47]. There were no dose-limiting toxicities and hence the maximum tolerated dose was not achieved. All enrolled patients could be scaled up to 24 mg/kg of birtamimab, which was established as the phase II recommended dose (Table 3). Furthermore, there were no clinical manifestations attributable to hypersensitivity, and no anti-birtamimab antibodies could be detected. The terminal elimination half-life of birtamimab in serum was between 13 and 16 days (Table 1), with a pharmacokinetic profile consistent with dosing by intravenous infusion every 28 days across the dose range. This is in stark contrast with the very short half-life of dezamizumab that was attributed to the sequestration of this antibody by the amyloid deposits. The reason for this discrepancy between two antibodies that are both supposed to target amyloid deposits has not been clarified. Exploratory secondary endpoints also included correlation studies of birtamimab exposure levels with changes in biomarkers of amyloid organ dysfunction. Cardiac and renal responses, defined in agreement with consensus, biomarker-based criteria [21], were seen in 57% and 60% of evaluable patients, respectively. While the study was not designed to assess efficacy and did not include a comparative control arm, these results were considered to compare favorably with respect to historical data sets [47].
Based on these positive results, two multicenter, randomized, double-blind, placebo-controlled, two-arm, efficacy and safety studies were started with birtamimab 24 mg/kg every 28 days as the experimental arm. The PRONTO study (NCT02632786) was an international, phase IIb study including AL patients who had at least a partial hematologic response to previous anti-plasma cell therapy and have persistent cardiac dysfunction (but NT-proBNP < 5000 ng/L) (Table 2). The primary endpoint of the study was the rate of cardiac response, while secondary endpoints included, among others, the rate of kidney and liver best response and functional measures. A total of 129 patients were randomized and could be evaluated (66 in the birtamimab arm and 63 in the placebo arm). There was no significant difference in the occurrence of serious adverse events in the two treatment arms. In an intention-to-treat analysis, there was no statistically significant difference in cardiac response, which was reported in 26/66 (39.4%) patients in the birtamimab arm and 30/63 (47.6%) patients in the placebo arm (p = 0.319) (Table 3). None of the prespecified, secondary endpoint analyses showed any signs of activity of the experimental drug, including renal and liver responses in evaluable patients, the physical component score of the Short-Form 36 (SF-36) questionnaire, and the 6MWT [48].
The VITAL study (NCT02312206) was a global phase III trial including newly diagnosed AL amyloidosis patients with cardiac involvement who were concomitantly receiving bortezomib-based frontline therapy (with a number of cycles of frontline therapy and eventual further lines of treatment at the discretion of the treating physician, as per standard of care) [49]. Patients eligible for and planning to undergo autologous stem cell transplant were excluded. The primary endpoint of this trial was time to composite of all-cause mortality or cardiac hospitalization, while the secondary endpoints included heart, kidney, and liver best response, functional measures, and patient-reported quality-of-life data (Table 2). Treatment was administered every 28 days until study completion, as this was an event-driven study. A total of 260 patients (130 patients per treatment arm) were enrolled. This study (along with other clinical trials on birtamimab that were open at that time) was terminated by the sponsor based on a futility analysis, whose results were disclosed shortly after the results from the PRONTO study. Death or cardiac hospitalization occurred in 56/130 (43.1%) patients in the birtamimab arm and 62/130 (47.7%) patients in the placebo arm, and there was no statistically significant difference in the composite primary endpoint (log-rank test: p = 0.3300; hazard ratio [HR] 0.835, with two-sided 95% confidence interval [CI] 0.5799–1.2011). Again, no specific safety signal emerged during the study, with similar rates of serious adverse events in the two study arms (Table 3). Data on secondary outcome measures have not yet been reported.
Based on a non-prespecified subanalysis on patients presenting with advanced amyloid disease (Mayo stage IV [50]) at baseline, showing a significant survival benefit favoring birtamimab in these patients, with 74% of birtamimab-treated patients alive at 9 months versus 49% of patients in the control group, a confirmatory, multicenter, global, randomized, double-blind, placebo-controlled phase III trial (the AFFIRM-AL study, NCT04973137) was started in patients with Mayo stage IV [51, 52]. In this study, birtamimab (24 mg/kg) or placebo will be administered every 28 days to newly diagnosed, Mayo stage IV AL amyloidosis patients receiving standard-of-care, bortezomib-containing therapy (± the anti-CD38 monoclonal antibody daratumumab at the discretion of the investigator). Patients with NT-proBNP levels exceeding 8500 ng/L or concomitant multiple myeloma are excluded from this trial. A total of 150 patients (100 in the birtamimab arm and 50 in the placebo arm) are planned to be enrolled. The primary endpoint of the study is time to all-cause mortality, while functional measures (the physical component part of the SF-36 questionnaire and the 6MWT) will be evaluated as secondary endpoints. Of note, the sponsor has reached a special protocol assessment (SPA) agreement with the FDA at fixing a p-value ≤ 0.10 (instead of the canonical 0.05) as the prespecified, significance threshold for the statistical analysis of the study outcome [52]. The study was started at the end of August 2021 and the estimated study completion date is June 2024. Results from this study are eagerly awaited.
AnselamimabThe monoclonal IgG1 antibody (mAb) anselamimab (CAEL-101) is the chimeric form of murine mAb 11-1F4, which was developed by Solomon’s group back in 2003 [53]. The antibody was produced by injecting, in BALB/c mice, the Vκ fragment derived from endopeptidase cleavage of a κ4 Bence Jones protein [54].
Subsequent studies indicated that the mAb 11-1F4 bound to a conformational neoepitope within the first 18 amino acids of misfolded human immunoglobulin LCs [53, 55], while it did not recognize native, circulating LCs. In vivo, the mAb expedited dissolution of AL amyloidomas in mice through the recruitment of neutrophils and activation of macrophages [54].
The specificity of the mAb was confirmed via immunohistochemical and radiographic detection by PET/CT of human AL amyloid deposits in patients who received the mAb intravenously [56].
These results led the National Cancer Institute’s Biological Resource Branch to production of the anti-amyloid fibril chimeric IgG1 antibody anselamimab (CAEL-101). The outcome of the phase Ia/b trial (NCT02245867) has been recently reported [57]. This was an uncontrolled, open-label, single-center, ‘up-and-down’ dose-escalation study investigating the tolerance, safety, pharmacokinetics, and possible clinical benefit of anselamimab (Table 2). Patients with measurable, localized, or systemic AL amyloidosis with relapsed/refractory disease were eligible. Dialysis, Eastern Cooperative Oncology Group (ECOG) performance status >3, and seriously limited cardiac, renal, or hepatic function were among the key exclusion criteria for this study. Single-patient cohorts were planned to receive either single infusions (phase Ia) or four weekly infusions (phase Ib) of the study drug starting at 0.5 mg/m2 and progressively increasing up to 500 mg/m2. If a patient experienced dose-limiting toxicity, then two additional patients were enrolled at the same dose, and the maximum tolerated dose was then defined as the highest dose at which two patients had no dose-limiting toxicity.
In the phase Ib study, the two highest doses were planned to be expanded to investigate dose response. A total of 27 patients were enrolled, all with systemic AL amyloidosis (8 in phase Ia and 19 in phase Ib). The median time since exposure to the last anti-plasma cell therapy was 2.6 and 7.4 months (range 0–15.5 months) in phases Ia and Ib, respectively. Of note, 5/8 patients in phase Ia and 15/19 patients in phase Ib had achieved at least a very good partial response or better to the anti-plasma cell therapy. No dose-limiting toxicity was registered up to 500 mg/m2 and the maximum tolerated dose was not achieved in either phase Ia or Ib. Grade 3 or higher adverse events included pruritus and pericardial effusion, reported in one patient each. One patient developed a mild, transient, and spontaneously resolving rash 4 days after a single dose of anselamimab 50 mg/m2, and a skin biopsy revealed a previously undiagnosed cutaneous amyloidosis with amyloid deposits that proved positive for the 11-1F4 antibody. Pharmacokinetic studies indicated a biphasic disposition of the antibody due to a rapid distribution phase, slower elimination phase, and long half-life of 10–16 days, consistent with the pharmacokinetics of other IgG antibodies, but in contrast with the 4–16 h half-life of dezamizumab (Table 1). Excluding three patients without measurable disease, the overall organ response rate was 63%, with a median time to response of 3 weeks [57]. Organ responses were noted only among evaluable patients receiving > 5 mg/m2 of anselamimab. A cardiac response was detected in 8/12 evaluable patients (67%) and was accompanied by a statistically significant improvement in global longitudinal strain, while the remaining patients had stable cardiac disease. A renal response was observed in 2/10 evaluable patients (20%), while 60% had stable disease and 20% experienced renal progression (Table 3). The organ response should be interpreted with caution because 7/8 patients with cardiac response and 1/2 patients with renal response had obtained a deep hematological response from previous chemotherapy, and a concurrent benefit on organ function due to profound reduction of the proteotoxic LC cannot be ruled out.
Anselamimab is under further clinical development, with a phase II (NCT04304144) trial and, notably, two phase III trials (NCT04512235 and NCT04504825) in patients with advanced amyloid cardiac involvement. The outcome of these two trials will be extremely informative on the possibility to recover organ function in patients who present with advanced organ damage.
Targeting the Amyloid Deposits in ATTR AmyloidosisPRX004Potential therapeutic amyloid-directed antibodies have been developed by several groups using a rational, structure-based approach, targeting epitopes that are inaccessible in TTR tetramers and exposed in the monomers or non-native conformations of TTR. This feature allows the targeting of amyloidogenic TTR species while preserving the functions of the native protein [58,59,60]. Using this rationale, structure-based, approach to generate mAbs that selectively targeted a TTR cryptotope, composed of residues 89–97, four mAbs were produced, showing high affinity and specificity for misfolded TTR species [60]. Mice were immunized with the TTR peptide 89–97 and screened for specific binding to non-native TTR conformations, suppression of in vitro TTR fibrillogenesis, promotion of antibody-dependent phagocytic uptake of misfolded TTR, and specific immunolabeling of ATTR amyloidosis patient-derived tissue. Four antibodies were selected, and the possibility to exploit these antibodies for the diagnostic imaging of amyloid deposits was also envisaged [60].
One of the four murine moAbs (which one was never disclosed by the Pharma company) was humanized and tested as PRX004 in an uncontrolled, international, multicenter, phase I, dose-escalation study (NCT03336580) assessing safety, tolerability, pharmacokinetics/dynamics, and immunogenicity of PRX004 (Table 2) [61]. The target population was represented by patients with ATTRv amyloidosis. Concomitant treatment with the TTR tetramer stabilizers tafamidis or diflunisal was permitted, provided that the dosing was stable in the previous 6 months, while prior or planned liver transplantation or recent exposure to TTR-silencing agents patisiran or inotersen was not allowed. This trial included a dose-escalation phase with a 3 + 3 design, exploring the range of up to three infusions of PRX004 at doses of between 0.1 and 30 mg/kg (at 28-day intervals), followed by an expansion phase and a long-term extension phase for up to 15 infusions. A total of 21 ATTRv patients completed the dose-escalation phase and 17 patients were enrolled in the long-term extension phase. No treatment-related serious adverse events occurred. Adverse events occurring in more than 10% of treated patients included fall, anemia, upper respiratory tract infection, back pain, constipation, diarrhea, and insomnia. The pharmacokinetics profile of PRX004 was consistent with IgG1 monoclonal antibodies. Although the study was not designed to assess efficacy, preliminary efficacy data were reported for seven evaluable patients (pooling data from 3, 10, and 30 mg/kg dosing groups under the assumption that dose levels ≥ 3 mg/kg should in principle enable clearance of > 90% of amyloid deposits). An improved global longitudinal strain (mean change: − 1.21%) was reported in all seven evaluable patients and an improved NIS was seen in 3/7 cases (Table 3). All seven evaluable patients were reported to have a slowed disease progression compared with natural history data.
A phase III study is under planning by the new pharma company that continued the development of the drug [62].
留言 (0)