
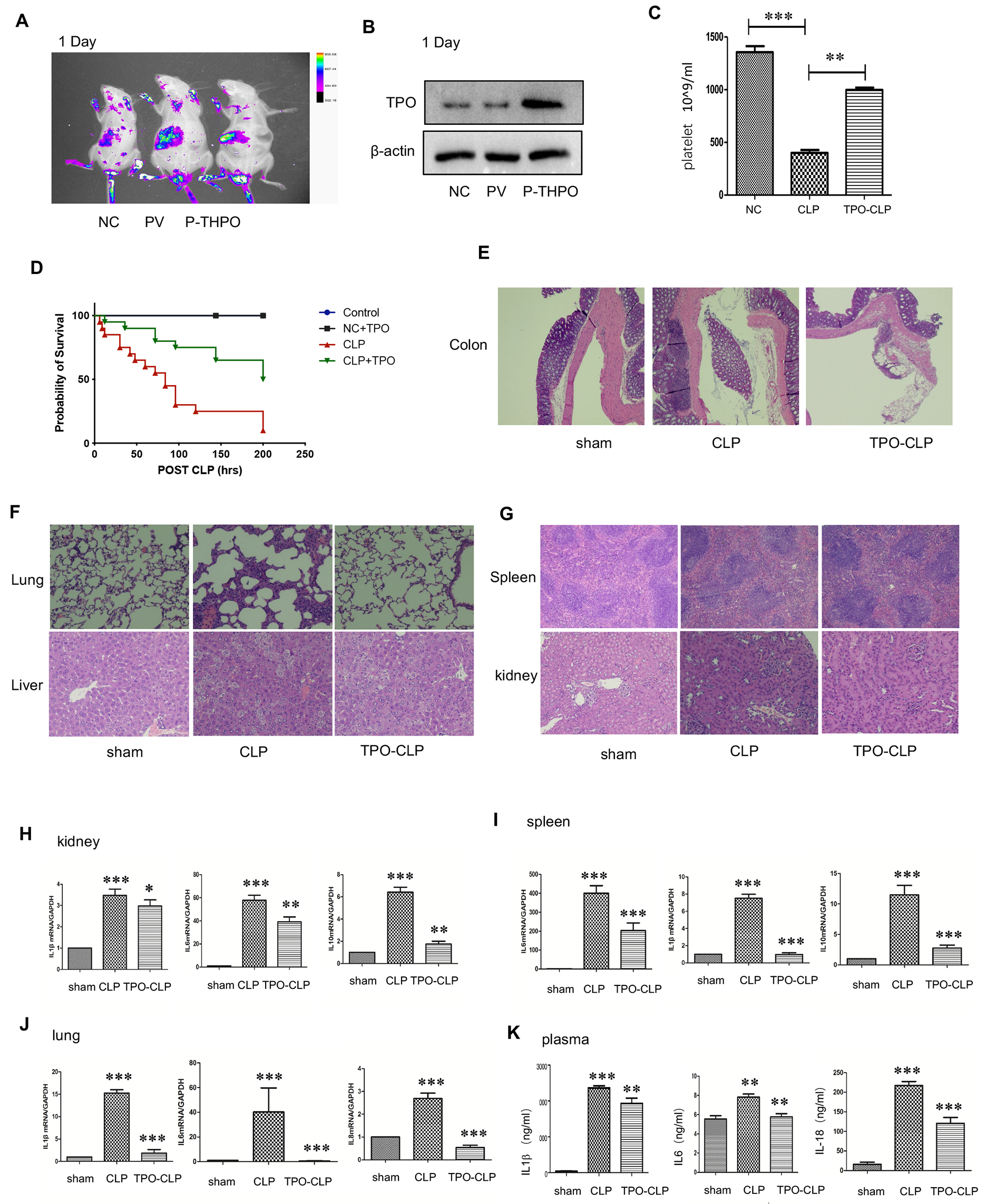
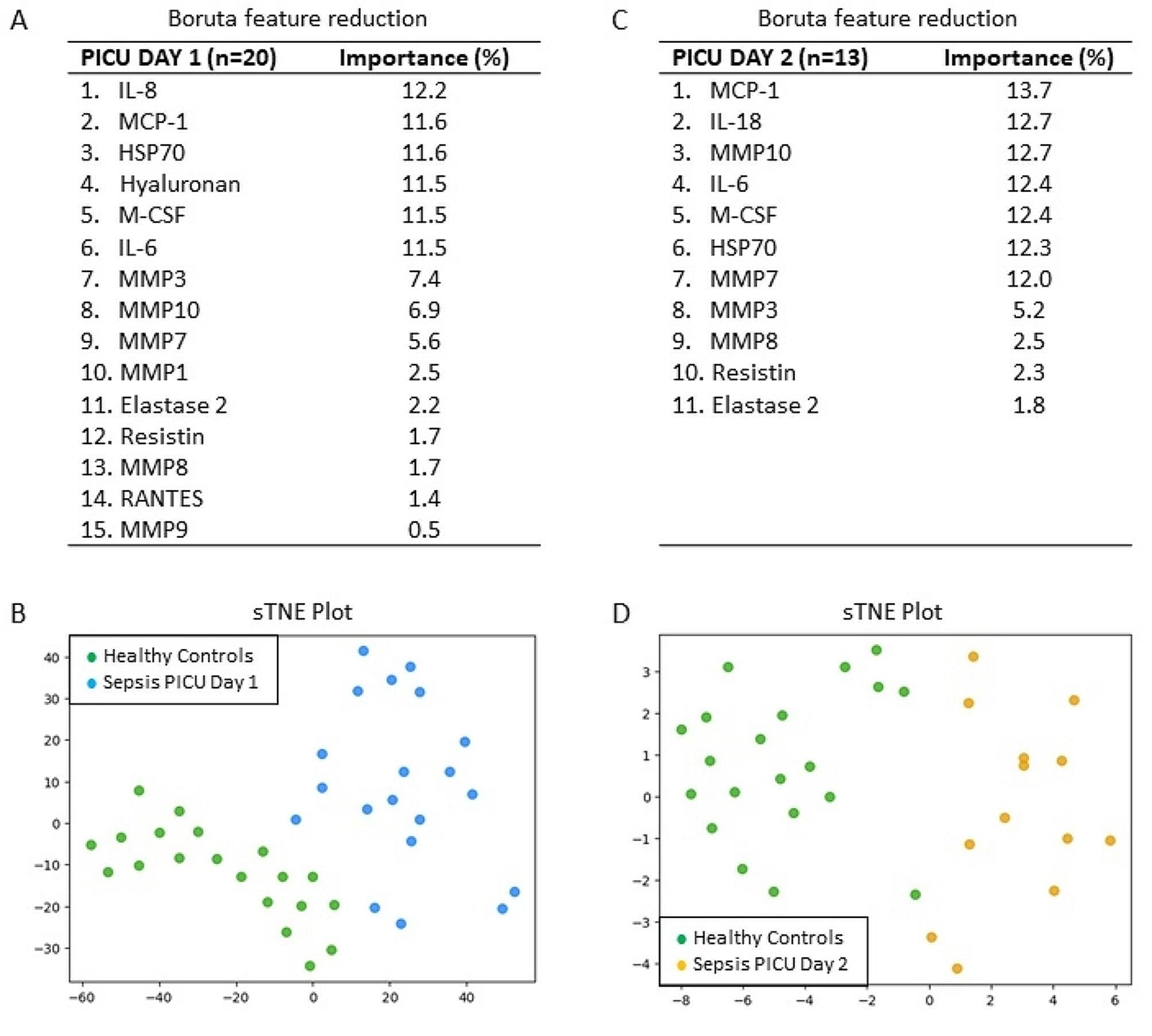
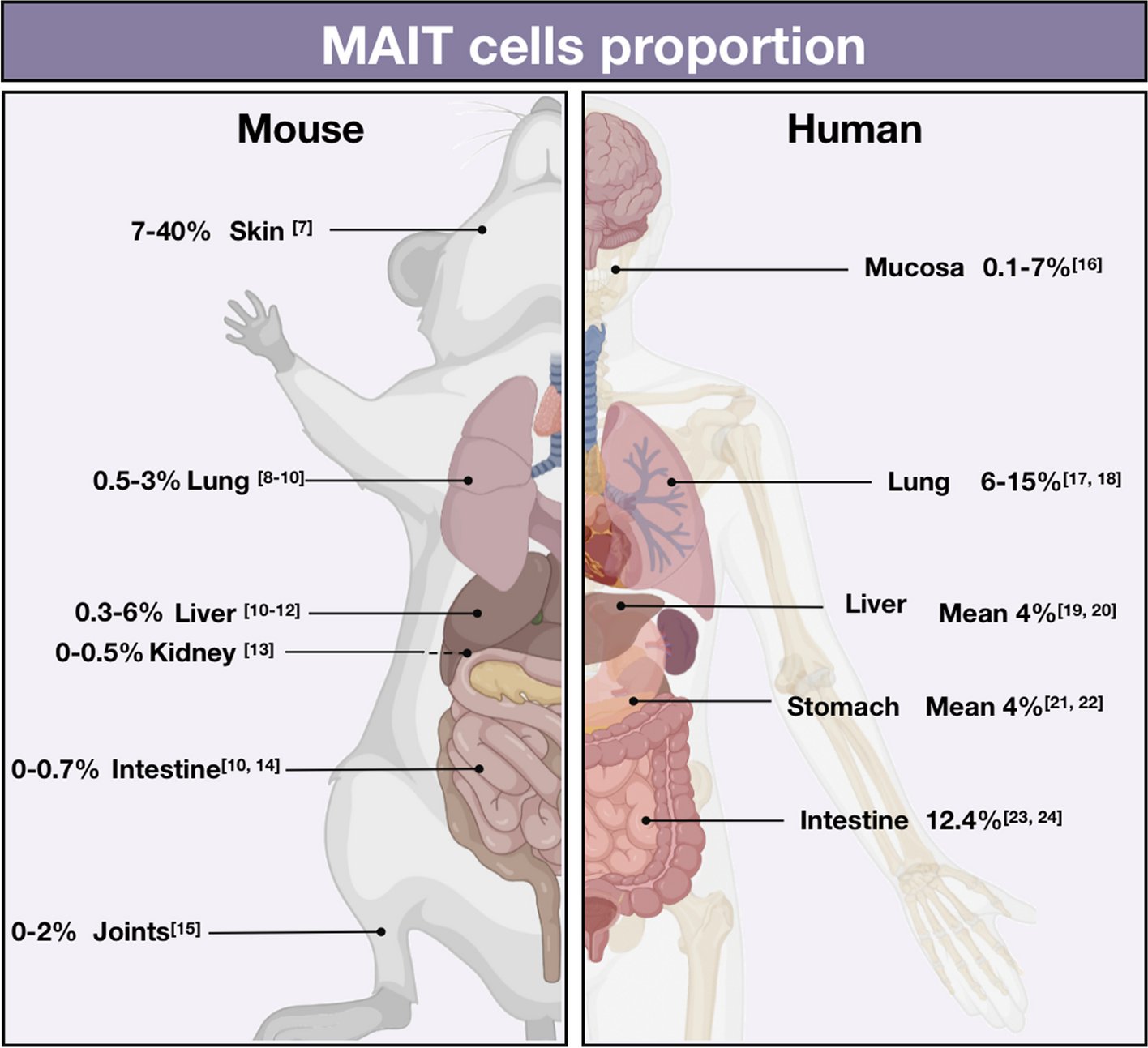
記住我




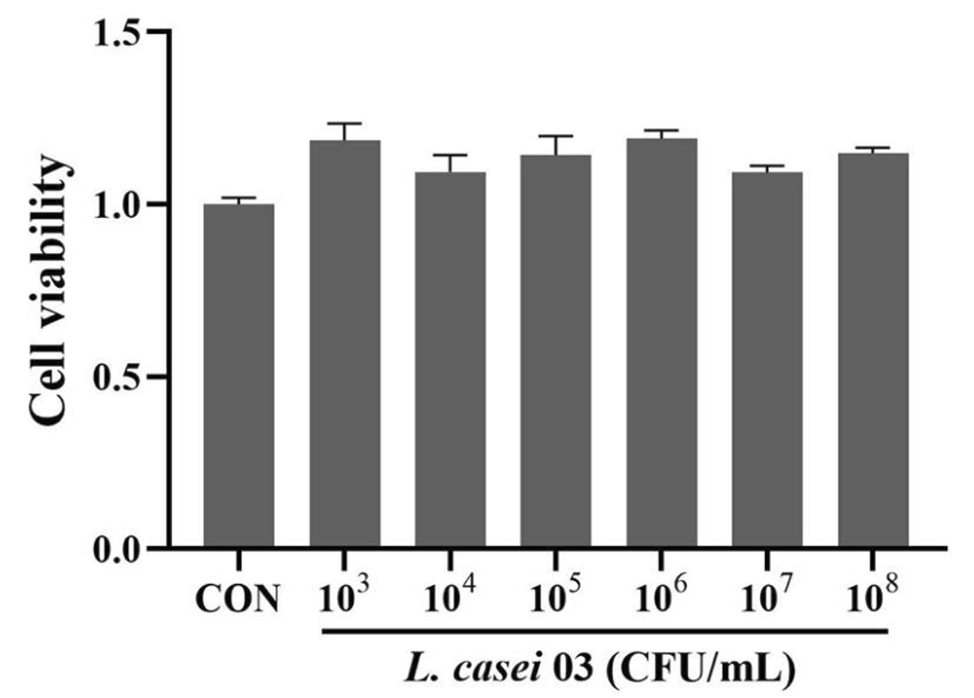
The epithelial cells in the BaLF samples from the M group of moderately infected SARS-CoV-2 patients, the S group of severely infected SARS-CoV-2 patients, and the control group (HC) of healthy people were divided into groups. The differences between the epithelial cells in the three groups were compared. The results showed that all epithelial cells were divided into 11 sub-clusters (Fig. 1A). The distribution of epithelial cells in the three groups (HC, M, and S) showed that epithelial cells were basically derived from the lavage fluid of Group S, while there were few epithelial cells in the lavage fluid of Group HC and Group M (Fig. 1B). The distribution of subsets of epithelial cells in the 13 samples is shown in Fig. 1C. The distribution of cells in the epithelial subsets of each sample and the corresponding cell numbers are detailed in Table 1. It can be seen that with the progression of SARS-CoV-2, the number of shedding epithelial cells in the alveolar lavage fluid increased. The situation of epithelial cells was further explored, and the expression of ACE2 gene in these epithelial cells was demonstrated. The results showed that ACE2 had relatively high expression in clusters 1, 3, and 7, and a small expression in clusters 4 and 5 (Fig. 1D). The histogram of the proportion of ACE2 gene expression in epithelial cells (Fig. 1E) shows that there were certain differences in the number, clustering, and positive rate of ACE2 expression between group M and group S in epithelial cells. In group S, the proportion of ACE2 positive cells was relatively high, which was consistent with the argument that ACE2 expression was the target of virus attack. To further understand the gene and functional characteristics of each cluster, we first used dot plot and violin plots to display the average expression and density distribution of the top1marker gene in the single-cell dataset clusters 1–11 of the cell-type cluster (Figs. 1F and H). In addition, a heat map was constructed between clusters 1–11 and the gene expression modules with clear differences (Fig. 1G). Among them, clusters 1, 3, 4, 5, and 7 highly expressed CHP2, ECM1, SLC23 A1, C1QB, and CRLF1. Clusters 2 and 6 highly expressed TRIB3 and IGLV3-19, and clusters 8, 9, 10, and 11 highly expressed MUC5B, SFTPA1, NUF2, and AC107959.4.
Fig. 1
Epithelial cell clusters in bronchoalveolar lavage fluid in patients with SARS-CoV-2 disease. A The 11 BaLF epithelial cells types. B Distribution of 11 BaLF epithelial cells types in healthy controls (HC) and patients with moderate (M) and severe (S) SARS-CoV-2 infection. C Component of epithelial cells in each sample. D Expression of ACE2 in epithelial cells in each group. E Histogram of ACE2 gene expression in epithelial cells. F Top marker gene dot plot of clusters 1–11. G Heat map of differential genes in cell-type clusters. H Violin plots showing distribution of expression for selected cluster marker genes.
Table 1 The number, cluster distribution and proportion of epithelial cellsDifferent functions of epithelial cells in patients with moderate and severe SARS-CoV-2 infectionTo further understand the differences in epithelial cells between group S and group M, difference analysis and enrichment analysis of the corresponding gene function were performed on the epithelial cells of group S and group M. The GO functional enrichment analysis showed that the up-regulated genes in group S were mainly related to the regulation of cell migration, defense response to the virus, and response to the virus and type I interferon signaling pathway (Fig. 2A). The KEGG signaling pathway analysis results showed that the up-regulated genes in the S group were mainly enriched in the apoptosis, IL-17 signaling pathway, fluid sheer stress atherosclerosis, and TNF signaling pathway (Fig. 2B). Both GO and KEGG showed that the epithelial cells in group S were inflammatory.
Fig. 2
Differences in the function of epithelial cells in patients with moderate and severe SARS-CoV-2 infection. A Gene Ontology (GO) enrichment analysis of the up-regulated genes in group S vs. group M. The size of the black spots indicates the gene number. B The KEGG of the up-regulated genes in group S vs. group M. The color gradient represents the P value, and the size of the dots represents the gene number. Gene Set Enrichment Analysis (GSEA) of the up-regulated genes in group S vs. group M, such as cytokine signaling in immune system (C), the chemokine signaling pathway (D), signaling by interleukin (E), and Toll receptor signaling pathway (F)
GSEA enrichment analysis was performed on the epithelial cells of S and M groups. Similarly, it was found that cytokine signaling in the immune system, signaling by interleukins, the chemokine signaling pathway and Toll receptor, and other signaling pathways were enriched. Most signaling pathways were closely related to inflammation activation (Fig. 2 C–F).
Interrelationships and functional changes among clusters of epithelial cell types expressing ACE2Owing to the relatively high expression of ACE2 in clusters 1, 3, and 7 and a small amount in clusters 4 and 5, the relationship among clusters 1, 3, 7, 4, and 5 cell populations with ACE2 expression was explored by the pseudo-time analysis method. The results showed that clusters 4 and 5 were ‘differentiated’ gradually through 7 to 1 and 3, suggesting that the epithelial cell populations of 7, 1, and 3, which were mainly concentrated with group S and expressed ACE, might have been attacked by the virus and subsequently changed, and 7 was an intermediate transition state (Fig. 3 A, B). This indicates that the SARS-CoV-2 virus may first recognize the ACE2 of the epithelial-cell-type clusters, clusters 4 and 5, and may then transform the epithelial-cell-type clusters into clusters 7, 1, and 3.
Fig. 3
Interrelationships and functional changes among clusters of epithelial cell types expressing ACE2. A The pseudo-time trajectory of epithelial cell clusters 1, 3, 4, 5, and 7. Color from dark to light, indicating the differentiation time from early to late. B Pseudo-time trajectory of cell population differentiation in epithelial cell clusters 1, 3, 4, 5, and 7, which could be used to infer the differentiation relationships among cell populations at the initial step. C Venn diagram showing that the number of differences in cluster 7 vs. 5 was 48 up-regulated genes. D Venn diagram showing 10 down-regulated genes in cluster 7 vs. 5. E KEGG analysis of the Venn diagram intersection of 48 up-regulated genes. F KEGG analysis of the Venn diagram intersection of 10 down-regulated genes. G Gene heat map analysis of clusters 1, 3, 4, 5, and 7. Red indicates higher gene expression. Blue indicates lower gene expression. H GSVA bar chart of cluster 1 vs. cluster 5. I GSVA bar chart of cluster 3 vs. cluster 5. Green indicates no significant difference. Blue indicates significant difference. A T value greater than 0 denotes an up-regulated pathway, while a T value less than 0 denotes a down-regulated pathway
Because cluster 5 was the main cell population in the HC group and was in the early stage of the pseudo-time analysis, while clusters 1, 3, and 7 were mainly in group S, the Venn difference analysis method was used to consider cluster 5 as the control. Clusters 1 and 3 and 7 epithelial subsets and cluster 5 were compared with each other. The Venn diagram showed that the number of differences between clusters 7 and 5 was relatively small, with 48 up-regulated genes (Fig. 3 C) and 10 down-regulated genes (Fig. 3 D). Cluster 7 was an intermediate transition state, and the number of differences was relatively small. Venn diagram intersection of up-regulated KEGG genes was mainly concentrated in the p53 signaling pathway, pathogenic escherichia coli infection, and IL-17 signaling pathway (Fig. 3 E); Venn diagram intersection of down-regulated KEGG genes was mainly concentrated on phagosome, antigen processing and presentation, and oxidative phosphorylation (Fig. 3 F). The results showed that clusters 1, 3, and 7 not only showed inflammation but also a certain degree of apoptosis.
In addition, we selected genes related to inflammation and compared the differences between clusters 1, 3, 4, 5, and 7 by drawing heat maps. The results showed that inflammatory genes were significantly expressed in clusters 1, 3, and 7, which indicated the activation of inflammatory pathways. The high expression of MUC5AC also indicated the increase of mucus secretion (PMID: 30352166, PMID: 30352166). However, the expression of inflammatory genes was lower in clusters 4 and 5 (Fig. 3 G).
To further confirm that clusters 1 and 3 have the common characteristics of activating inflammatory pathways, GSVA analysis was conducted on clusters 1 and 3. Compared with cluster 5, the results showed that cluster 1 was mainly enriched in the FoxO signaling pathway, human papillomavirus infection, TNF, and IL-17 signaling pathway (Fig. 3 H). Similarly, compared with cluster 5, cluster 3 was mainly enriched in the FoxO signaling pathway, ErbB signaling pathway, AMPK, TNF, and IL-17 signaling pathway, among others (Fig. 3 I). Clusters 1 and 3 showed activation of inflammatory pathways.
Functional changes of clusters of epithelial-cell types that did not express ACE2 after infection with SARS-CoV-2The expression of clusters 2 and 6 was concentrated in group S, but the expression data showed that there was almost no expression of ACE2 in these clusters. To understand the change of clusters 2 and 6, KEGG enrichment analysis of differential genes of clusters 2 and 6 was conducted (Top 10). The results showed that, compared with cluster 5, many up-regulated genes of clusters 2 and 6 were related to inflammation, suggesting that clusters 2 and 6 might be related to clusters 1, 3, and 7, which might also be affected by the virus. Cluster 2 showed a strong apoptotic function, while cluster 6 did not show that ability (Fig. 4 A, B).
Fig. 4
Functional changes of clusters of epithelial cell types for which no clusters express ACE2 after infection with SARS-CoV-2. A KEGG enrichment analysis of differential genes of cluster 2 vs. 5. B KEGG enrichment analysis of differential genes of cluster 6 vs. 5. C GSVA bar chart of cluster 2 vs. 5. GSEA of the up-regulated genes in cluster 6 vs. 5 such as activation of immune response (D) and leukocyte-mediated immunity (E)
To further understand the relationship between clusters 2 and 6 and 1, 3, 7, 4, and 5, the relationship between these pathways was further studied. GSVA analysis showed that, compared with cluster 5, cluster 2 was mainly enriched in ubiquitin-mediated proteolysis, the TGF-beta signaling pathway, the TNF signaling pathway, and the human cell leukemia virus 1 infection signaling pathway (Fig. 4 C)—while, compared with cluster 5 epithelial cells, cluster 6 epithelial cells were enriched in the active immune response and leukocyte-mediated immunity signaling pathway (Fig. 4 D, E). Both the GSVA analysis and GSEA results of clusters 2 and 6 showed activation of inflammation and immune function after infection with SARS-CoV-2.
Interaction between clusters of different epithelial cell types after SARS-CoV-2 infectionThe previous results showed that clusters 2 and 6 were mainly found in group S, and the functions of clusters 2 and 6 also showed activation of immunity and inflammation. Thus, was there a certain degree of difference between cluster 2 and cluster 6, compared with clusters 1, 3, and 7? To find out, clusters 1, 3, 7, 2, 6, 4, and 5 were put together for the pseudo-time analysis (the relationships among 1, 3, 7, 4, and 5 are shown in Fig. 3). The results showed that clusters 4 and 5 were in the original state and differentiated in two directions—one was cluster 2, and the other included clusters 1, 3, and 6. Cluster 7 was an intermediate state (Fig. 5 A, B).
Fig. 5
Interaction between clusters of different epithelial cell types after SARS-CoV-2 infection. A The pseudo-time trajectory of epithelial cell clusters 1, 2, 3, 4, 5, 6, and 7. Color from dark to light, indicating the differentiation time from early to late. B Pseudo-time trajectory of cell population differentiation in epithelial cell clusters 1–7. C Similarity heat map between clusters 1–7. Similarity heat map between clusters 1–7. Red represents high similarity, and blue represents low similarity. D Fifty marker genes of clusters 1–7 were selected for heat mapping display. E UpSet diagram of the differentially expressed genes in clusters 1, 3, 7, 2, and 6. F KEGG signaling pathway analysis of up-regulated differential genes in epithelial cell cluster 2. G KEGG signaling pathway analysis of down-regulated differential genes in epithelial cell cluster 2. H Heat map of cell death gene expression module in epithelial cell clusters 1–7
To further determine the relationship between clusters 1, 2, 3, 4, 5, 6, and 7, similarity heat map analysis was conducted on these clusters. The results showed that clusters 1, 3, 6, and 7 had relatively high similarity (> 0.9), and clusters 4 and 5 were similar (similarity > 0.9). Cluster 2 was relatively specific, and the differences between cluster 2 and the other clusters were relatively large. This result was consistent with that of pseudo-time analysis. This suggested that although cluster 2 was also activated by inflammatory pathways compared with cluster 5, this cluster might have had its own special characteristics (Fig. 5 C). Fifty marker genes of clusters 1–7 were selected for mapping display. The results also showed a certain degree of similarity between clusters 1, 3, and 6, and cluster 2 had specificity (Fig. 5 D).
The results of the pseudo-time analysis showed that cluster 7 was a cell population that transitioned to clusters 1, 3, and 6, and cluster 6 was similar to clusters 1 and 3. Cluster 2 was another branch. Comparing the differences between clusters 1, 3, 7, 2, and 6 with cluster 5, the UpSet results showed that the differential genes of cluster 7 were indeed relatively few. Moreover, the intersection of the differential genes among several groups was strong, and cluster 2 had a large number of differential genes with specific changes (Fig. 5 E). This suggested that cluster 2 may have been different from the other clusters to some extent (and in the results of pseudo-time analysis, cluster 2 was also independently divided into one cluster, Fig. 5 A, B).
To clarify the functional characteristics of cluster 2, the specific differential genes of cluster 2 were differentiated, and the corresponding functional enrichment analysis was conducted. KEGG signaling pathway analysis results showed that the up-regulated differential genes of cluster 2 were mainly related to the VEGF signaling pathway, apoptosis, and necroptosis (Fig. 5 F). The down-regulated differential genes of cluster 2 were mainly enriched in the ribosome, protein processing in the endoplasmic reticulum, and endocytosis (Fig. 5G). The single-cell dataset clusters 1–7 of the cell-type cluster and the cell death gene expression module were used to construct a heat map. The results showed that many apoptotic genes in cluster 2 began to be highly expressed (such as BAD and TNFRSF10B), indicating that the apoptotic condition of cluster 2 was significantly higher than that of other clusters (the genes of clusters 1, 3, 7, and 4 were relatively higher than those of clusters 5 and 6) (Fig. 5 H).
The communication relationship between epithelial cell-type clusters and immune cellsTo understand the signal communication relationship between epithelial cells and immune cells, we used single-cell RNA sequencing data, took the gene expression data of cell subpopulations as the research object, used the ligand-receptor database, and used cellphoneDB to obtain information about ligands and receptors in cells to reflect the interactions between cells [12]. Figure 6 shows the communication relationship between the cluster 2 epithelial cells and the T, NK, and macrophages. Among them, MIF was highly expressed in the cluster 2 epithelial cells, and its receptor TNFSRSF14 was expressed in macrophages. The MIF secreted by the cluster 2 epithelial cells could interact with the receptor TNFSRSF14 in macrophages. In addition, the GRN secreted by macrophages could interact with the EGFR receptor of the cluster 2 epithelial cells. The expression pattern of the MIF-TNFSRSF14 and GRN-EGFR ligand-receptor complex interaction could accurately describe the interaction between the cluster 2 epithelial cells and macrophages. Similarly, the interaction between NK cells and cluster 2 epithelial cells, T cells, and cluster 2 epithelial cells could be accurately reflected (Fig. 6 A).
Fig. 6
The communication relationship between epithelial cell-type clusters and immune cells. A Circos diagram of cluster 2 and immune cells. The solid line in the figure indicates that the interaction relationship is significant. The first circle represents the expression level of the ligand/receptor gene; the higher the red expression level, the lower the green expression level. The second circle represents cell types, such as epithelial cell cluster 2, macrophages, T cells, and NK cells. The third circle represents the ligand (L)/receptor (R) to which the gene belongs, and the fourth circle represents the interaction relationship between the ligand-receptor gene pair. B Cell communication dot diagrams between clusters 1–7 and macrophages. C Cell communication dot diagrams between clusters 1–7 and T cells. D Cell communication dot diagrams between clusters 1–7 and NK cells. The row represents the ligand-receptor pair that has a communication relationship between cells. The column represents the cell type (receptor cell | ligand cell) where cell communication occurs. The red color of the circle represents the expression value of the interacting cell gene, that is, the redder the circle, the greater the expression value
Furthermore, we compared the communication relationship between different clusters of epithelial cells and macrophages, T cells, and NK cells. Similar findings, the ligand-receptor expression patterns of TNFRSF10D-TNFSF10, TNFRSF10D-MIF, and TNFRSF10B-TNFSF10, showed that the cluster 2 epithelial cell cluster was different from other clusters and had a special interaction with macrophages but not with T cells and NK cells (Fig. 6 B, C, D).
留言 (0)