
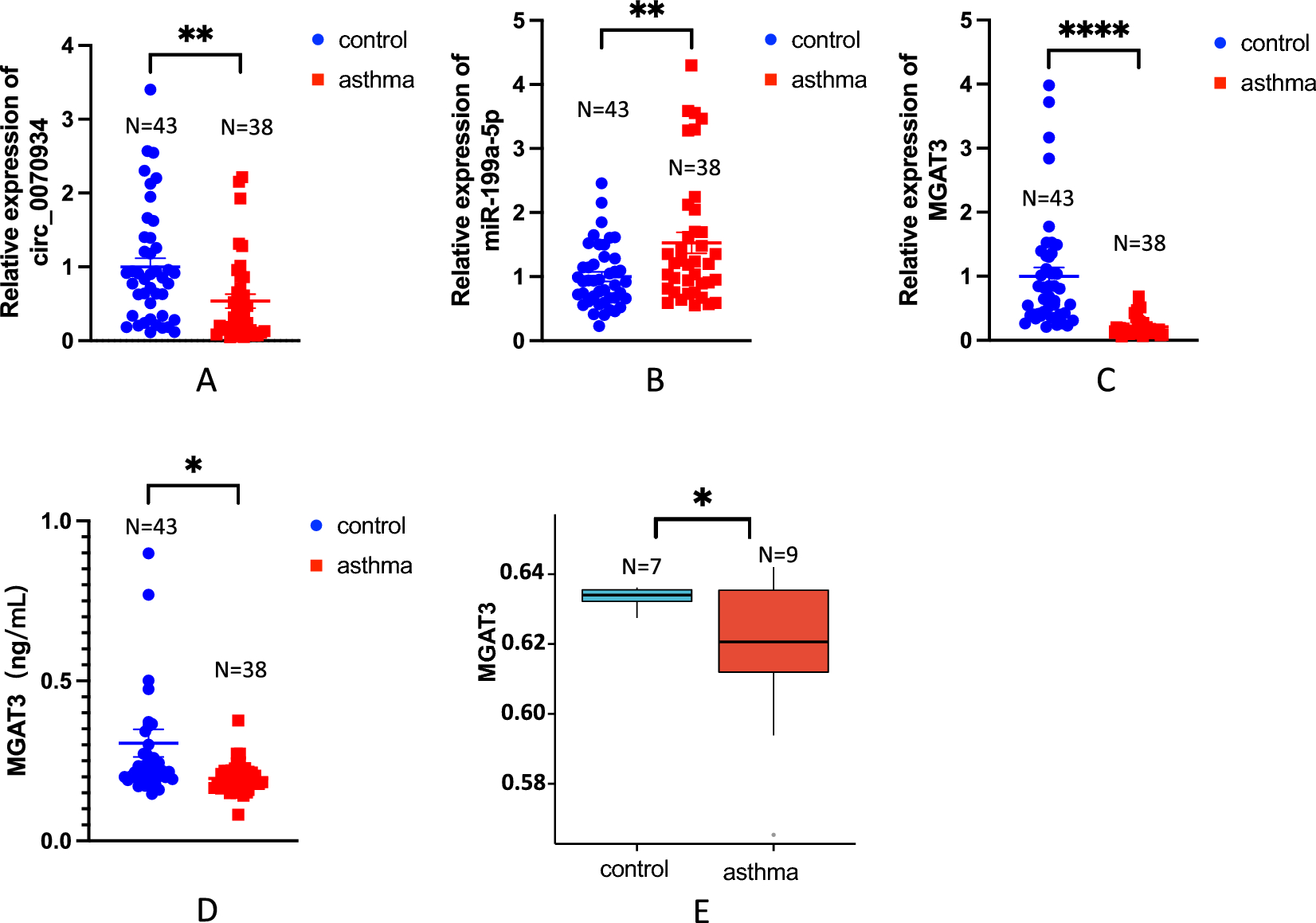
記住我

Differential diagnosis for skin rash with nodal involvement is broad including malignancy, disseminated MAC, and autoimmune process. The presence of enlarged histiocytes with foci of emperipolesis, or the engulfing of mature immune cells by histiocytes with abundant cytoplasm, initially raised the suspicion for a potential diagnosis of Rosai-dorfman disease [8]. However, typically in RDD immunohistochemical staining also shows expression of CD68 and S100, but not CD1a which we did not find in our patient [8]. There have also been several case reports of RDD diseases mimics with Rosai-Dorfman like histological features including non-tuberculosis mycobacterial infections [9]. Our patient’s skin biopsy was negative for acid fast bacteria making MAC infection less likely.
Rosai-Dorfman-like histological features can also be seen in IgG4-RD [10]. The discovery of hypergammaglobulinemia with elevated IgG4, characteristic histopathological findings and multi-organ involvement led to the diagnosis of IgG4-RD. Diagnostic criteria IgG4-RD was first established in 2011 by Umehara et al. (Table 1, Fig. 5) and later updated in 2020 to exclude solitary nodal disease as criteria for diagnosis [11]. Our patient met all 3 of the diagnostic criteria, with his uveitis thought to represent multiorgan involvement, making a definite diagnosis of IgG4-RD.
Table 1 Consensus diagnostic criteria for IgG4-RDFig. 5
Diagnostic algorithm for IgG4-RD
Major histopathologic features for extranodal IgG4-RD have been reported with increased specificity for IgG4-RD. These include a characteristic storiform pattern of fibrosis seen on histopathology as was seen intranodally with our patient [5, 12, 13]. However, various histologic patterns have been described in IgG4-related lymphadenopathy: Type I, multicentric Castleman disease-like; Type II, follicular hyperplasia; Type III, interfollicular expansion; Type IV, progressive transformation of germinal centers; and Type V, inflammatory pseudotumor-like. These pathologic findings are different than those seen in extranodal disease, with the exception of the type V pattern, and are relatively nonspecific.
Interestingly, a reactive dermatitis with neutrophilic infiltration (commonly seen as Sweet’s syndrome and erythema pustulosis) is also associated with AOID [2, 7, 14]. Our patient’s skin biopsy was positive for neutrophilic infiltration, and this, along with his pulmonary MAC infection, was an early and important diagnostic clue for an underlying diagnosis of AOID. One study found that the positive predictive value for AOID increased from 0.154 in patients with a diagnosis of NTM alone to 1.0 in patients with a diagnosis of NTM and a reactive dermatitis [15]. If seen in the future, this combination of disease should prompt investigation with an IFN-gamma autoantibody assay.
In a Thailand and Taiwan cohort, IFN-gamma auto-antibodies were detectable in over 80% of patients suffering from disseminated NTM without previously known risk factors such as human immunodeficiency virus (HIV) [2]. Diagnosis of our patient was eventually established by the presence of anti-IFN-gamma autoantibodies in the serum. This diagnosis was delayed, however, due to concomitant treatment of IgG4-RD that resulted in an initial false negative anti-IFN-gamma level. This reinforces the need for high clinical suspicion of immunodeficiency in a patient of Southeastern Asian descent with reactive dermatitis and an opportunistic infection without known risk factors. If there is such a suspicion, cessation of any immunosuppressive treatment should be considered for prompt diagnosis.
The relationship between IgG4-RD and AOID is an area that requires further research and has only been described twice before to our knowledge [6, 7]. The cause of anti-IFN-gamma autoantibody generation in AOID is unknown, but research suggests there may be a genetic component as there is an association with allele polymorphisms in human leukocyte antigens DRB1 and DQB1 [16]. IFN-gamma regulates the toll-like receptor pathway in response to intracellular pathogens. The disruption of this pathway and subsequent reactive oxygen species production, antigen presentation and interleukin 12 production leads to susceptibility to NTM disease such as MAC [1].
To date, the pathogenesis of IgG4-RD is also incompletely understood. Some have suggested that IgG4-RD is precipitated by a likely unknown antigen that causes B cell differentiation into IgG4 producing plasma cells leading to the increased expression of T helper 2 (Th2) cells and cytokines [17, 18]. The immune mediated response of IgG4-RD shares a similar response to tuberculosis infection as the activation of Helper T cells and the expression of similar cytokines in IgG4-RD have a crucial role in our immune mediated defense against tuberculosis infection [19, 20].
As disseminated mycobacterial disease has been shown to increase a Th2 immune mediated response, it has been suggested that prolonged opportunistic infections due to underlying AOID results in activation of Th2 cells and repeated antigen exposure causes increased IgG4 production [6, 21]. Another possible mechanism of overlap between the two diseases is due to the generation of anti-IFN-gamma autoantibodies itself. It was found that the level of anti-interferon-gamma IgG4 was disproportionately high compared to the total IgG subclass distribution, suggesting that the auto antibodies are excessively of IgG4 subtype [2].
Management strategies of both diseases involve steroids and biologic agents. International consensus guidelines recommend that all symptomatic patients with IgG4-RD and most asymptomatic patients with the potential for irreversible organ damage receive induction therapy due to the metachronus nature of the disease [5]. Induction therapy includes prednisone at a dose range of 30–40 mg daily for at least 2–4 weeks with a following taper that can range from 3 months to 3 years based on expert opinion [5]. Most experts also agree with the addition of a steroid sparing biologic agent to induce more durable remission with less relapse and to minimize toxicities from prolonged steroid exposure [5]. Options for biologic therapy include rituximab, cyclophosphamide, azathioprine, 6-mercaptopurine, mycophenolate mofetil, methotrexate, tacrolimus, and leflunomide.
Treatment for AOID is an area of ongoing research with no published guidelines to guide management choices. Multiple treatments have been trialed in case reports with varying success including intravenous immunoglobulin, subcutaneous IFN-gamma, plasmapheresis, cyclophosphamide and rituximab [3, 22,23,24,25,26]. One study featured a modified lupus nephritis protocol with 5–35 cycles of cyclophosphamide (400 mg) with adjunct steroid use and 10–48 months of antibiotic use and had remission with resolution of infection in 2 out of 7 patients, continued infection without hospitalization in 3 out of 7 patients, and relapsed disease and hospitalization in 2 out of 7 patients [22]. Another study infused rituximab according to a lymphoma regimen (375 mg/m2 weekly) for 9–15 cycles over 1–5 years in 4 patients with periods of relapsed disease but overall clinical, radiologic, and laboratory improvement [26]. Early data suggests that management requires both anti-microbial treatment as well as reestablishment of the IFN-gamma pathway through immunosuppressive therapy [26]. This presents a unique, clinical challenge as immunosuppressive treatment can prolong highly morbid infections.
The management of pulmonary MAC in general remains challenging, especially in an immunocompromised host. The advent of macrolide-based regimen was a turning point in MAC treatment, leading to its foundational recommendation in guidelines since 1997 [27,28,29]. The most recent 2020 guideline recommends a macrolide-based regimen over non-macrolide-based regimen based on extensive observational studies showing higher rates of culture-conversion and that macrolide susceptibility predicts treatment success [27]. It further recommends a three-drug over two-drug regimen to mitigate risk of emerging macrolide resistance and a duration of at least twelve months after culture conversion [27]. But treatment success remains essentially a coin toss with an overall success rate of 52.3% and up to only 61.4% in those adhering to guideline-based therapy [30].
While immunosuppression is an established risk factor for mycobacterial disease, robust evidence-based guidelines are lacking for most immunocompromised hosts, including patients with transplants [31] and those like our patient with a rare immunodeficiency. The best evidence comes from patients with HIV and disseminated MAC. Treatment recommendations essentially remain the same as those for immunocompetent patients, but the guideline recommends primary prophylaxis with a macrolide in those with CD4 counts less than 50 cells/mm3 and, specifically, not taking antiretroviral therapy (ART), replacing the prior recommendation to prescribe prophylaxis to all patients with CD4 counts less than 50 cells/mm3 regardless of ART [32]. This change reflects the cornerstone importance of reconstituting immunity as a means to treat and prevent mycobacterial diseases, a strategy we attempted in our patient.
The largest longitudinal study on AOID treatment found that infection clearance was dependent on the use of immunosuppressives [33]. The median time of infection clearance was 3 years with antibiotics alone, 4 years with antibiotics and rituximab, 5 years with antibiotics and cyclophosphamide [33]. We elected to pursue a 4 year course of antibiotic treatment in our patient as he had received 5 cycles of rituximab and 2 cycles of cyclophosphamide for his IgG4-RD treatment. However, by the time of AOID diagnosis, the patient’s disseminated disease was so pervasive that, despite optimal treatment, that patient was transitioned to comfort care.
This case highlights the diagnostic challenge of concomitant AOID, IgG4-RD and disseminated MAC infection, the severity of morbidity and reviews optimal treatment strategies. Because of the novelty and rarity of these diseases, further research is needed to help classify the spectrum of disease between AOID, IgG4-RD and disseminated MAC infection in order to refine treatment strategies in this growing, high-risk population.
留言 (0)