
記住我
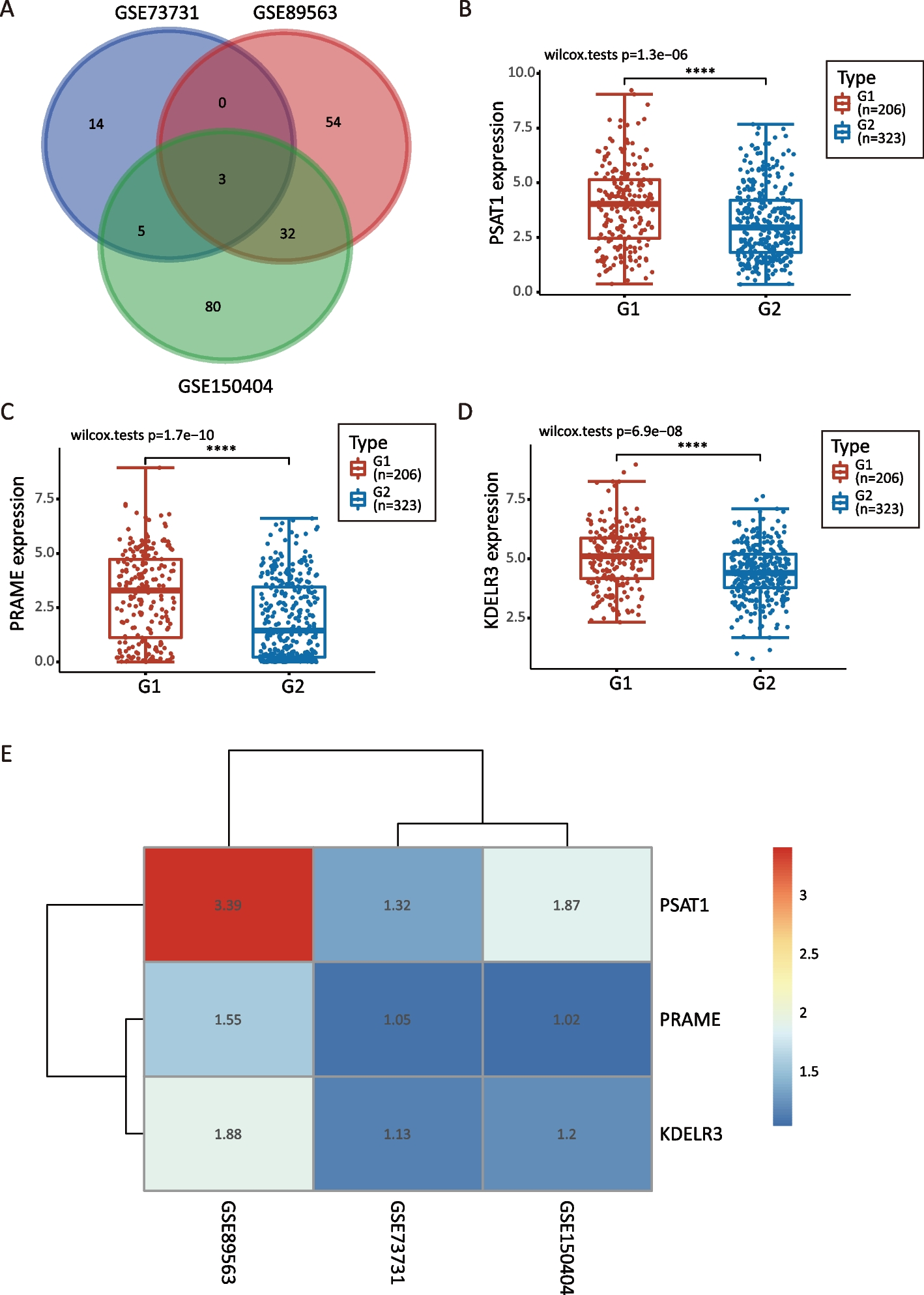
The trial, RESTART (LOGIK2002), is a multicenter, single-arm phase II study designed to investigate the efficacy and safety of carboplatin/nab-paclitaxel/atezolizumab combination therapy for individuals with advanced nonsquamous NSCLC and impaired renal function (Fig. 1). Eligible patients are registered prospectively.
Fig. 1
Design of the RESTART trial. NSCLC, non–small cell lung cancer; ECOG PS, Eastern Cooperative Oncology Group performance status; CCr, creatinine clearance; CNS, central nervous system; CBDCA, carboplatin; nab-PTX, nanoparticle albumin-bound paclitaxel; PD, progressive disease; RECIST v1.1, Response Evaluation Criteria in Solid Tumors version 1.1; ORR, objective response rate; PFS, progression-free survival; OS, overall survival; DOR, duration of response
Treatment planAtezolizumab is administered at 1200 mg on day 1 of consecutive 3-week cycles. Carboplatin is administered at an initial dose determined by Calvert’s formula to yield an area under the concentration-time curve of 5 mg mL− 1 min on day 1 of each 3-week cycle [14]. Nab-paclitaxel is administered at a dose of 100 mg/m2 on days 1, 8, and 15 of each 3-week cycle. After four cycles of induction therapy, maintenance therapy with atezolizumab is administered until disease progression, loss of clinical benefit, or development of unacceptable toxicity.
Eligibility criteriaIndividuals 20 years of age or older with histologically or cytologically confirmed nonsquamous NSCLC and impaired renal function (CCr of ≥15 but < 45 mL/min as calculated by the method of Cockcroft and Gault [15]) are eligible. (We decided to exclude patients with a CCr of < 15 mL/min because, in cases of chronic kidney disease, the introduction of maintenance hemodialysis is generally considered when the eGFR falls to ≤15 mL min− 1 1.73 m− 2.) Patients with sensitizing driver genetic alterations of EGFR, ALK, ROS1, BRAF, MET, RET, or NTRK are ineligible. Each patient is required to be at clinical stage III without indication for definitive thoracic radiotherapy, to be at stage IV, or to have recurrent disease after surgery or definitive radiotherapy that is not curable by local therapy. Patients must not have been treated previously with either cytotoxic chemotherapy or ICIs. The expression level for PD-L1 on tumor cells is not a determinant of eligibility. An Eastern Cooperative Oncology Group performance status of 0 or 1 as well as adequate lung, bone marrow, and liver function are required. Asymptomatic central nervous system metastases are permitted.
Patients are not eligible for the study if they have synchronous double or multiple cancers or have had metachronous double or multiple cancers within 2 years; have active hepatitis B or active hepatitis C or other infectious disease requiring systemic treatment; show obvious interstitial lung disease on chest computed tomography (CT); are receiving continuous systemic corticosteroid or immunosuppressant treatment; have other serious medical conditions including uncontrolled diabetes, unstable angina, and clinically serious arrhythmia; manifest peripheral neuropathy of grade ≥ 2; have hypersensitivity to carboplatin, nab-paclitaxel, or atezolizumab or to formulation additives; are affected by a psychological disorder that makes it difficult to participate in the study; or are pregnant, within 28 days after parturition, or breast feeding.
Evaluation of response and safetyA CT or magnetic resonance imaging scan of the brain, CT scans of the chest and abdomen, and a bone scan or positron emission tomography scan are required before onset of the study treatment. Contrast agents are not needed for the imaging tests. Patients undergo tumor assessment at baseline, every 6 weeks during the first 24 weeks, every 9 weeks for the next 27 weeks, and every 12 weeks thereafter. Tumor response is evaluated in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1). Adverse events are recorded based on the National Cancer Institute Common Terminology Criteria for Adverse Events (version 5.0).
Study endpointsThe primary end point is confirmed objective response rate, with secondary end points including progression-free survival, overall survival, duration of response, duration of protocol treatment, dose intensity, relative dose intensity, and safety. Subgroup analysis is stipulated for patients with a CCr of 15 to 30 mL/min or 30 to 45 mL/min, and the safety of CnP-atezolizumab will be evaluated with close monitoring for patients with a CCr of 15 to 30 mL/min.
Statistical considerationsThe primary end point is confirmed objective response rate. The expected response rate of this regimen in advanced nonsquamous NSCLC patients with impaired renal function is assumed to be 49.2%, which corresponds to that for the CnP-atezolizumab group in the IMpower130 study [2], and the threshold response rate is assumed to be 31.9%, which corresponds to that for the CnP group in the same study. The necessary sample size is estimated to be 35 subjects, with a one-sided significance level (α value) of 10% and a power of 80%. Taking into account ineligible subjects and those lost to follow-up, the target sample size was determined to be 40 subjects.
留言 (0)