
記住我
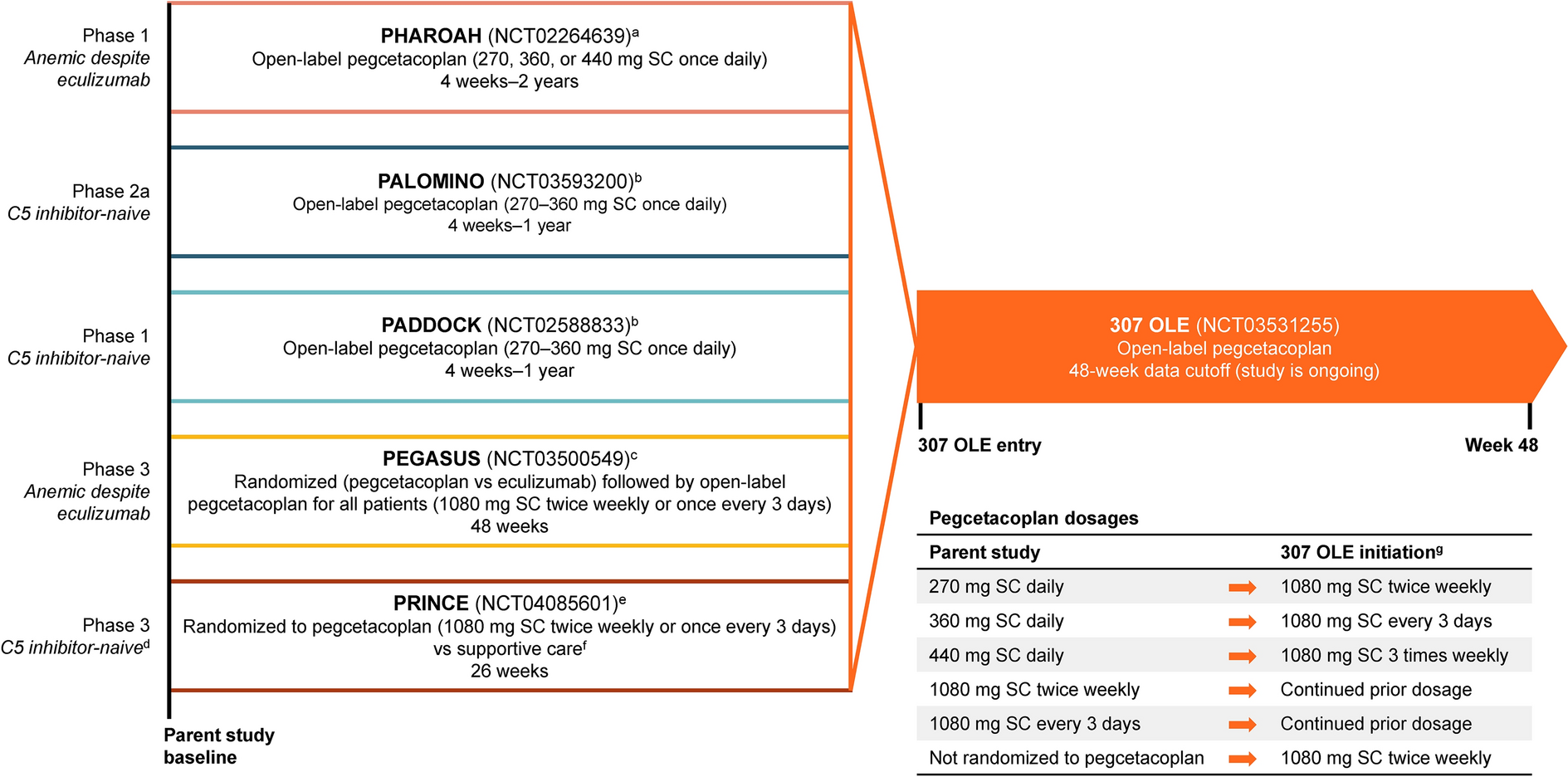


This was a randomized, double-blind, placebo-controlled, phase 1 study that was conducted sequentially in two parts, with two cohorts in each phase (four cohorts in total). The primary objective was to evaluate the safety and tolerability of single and multiple doses of TPIP, and the secondary objective was to evaluate the PK of TP and treprostinil in healthy volunteers. PPD Phase I Clinic (Austin, TX, USA) generated the randomization schedule, and each cohort was independently assigned by a qualified individual who was not involved in study conduct, data management, or data analysis. All participants entered the study after a 27-day screening period.
In the single-dose phase, the first cohort of participants was randomized 1:1:1 to receive an inhaled dose of TPIP (112.5 µg, 225 µg, or 450 µg), and the second cohort of participants was randomized 3:1 to receive TPIP 675 µg or placebo (Fig. 1). Participants were admitted to the clinic for 4 days, during which a single dose of assigned treatment was administered on day 1 followed by 3 days of assessments and observations. Safety data were collected for 72 h after dosing. A blinded safety review was conducted before dose escalation. On the basis of the review of safety data, the sponsor and investigator could have chosen to repeat a dose level, administer a dose higher or lower than the previous dose, escalate to a dose that was higher or lower than the next planned dose, and/or change the number of participants to be randomized to receive placebo. Participants were discharged on day 4 and contacted via telephone for a safety follow-up approximately 30 days after dosing.
Fig. 1
Study design. All doses were administered using 112.5-μg single-actuation capsules. d day, PK pharmacokinetics, QD once daily, Scn screening, TPIP treprostinil palmitil inhalation powder. aBlood samples for PK assessments in the single-dose phase were collected within 15 min before dosing and at 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24 (day 2), 36 (day 2), 48 (day 3), and 72 (day 4) hours after administration of TPIP or placebo; blood samples for PK assessments in the multiple-dose phase were collected within 30 min before dosing and at 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, and 12 h after dosing on day 1, before dosing only on days 2, 3, 4, 5, and 6, and before dosing on day 7 and 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24 (day 8), 48 (day 9), and 72 (day 10) hours after day 7 dosing
In the multiple-dose phase, the first cohort of participants was randomized 3:1 to receive an inhaled dose of TPIP 225 µg once daily (QD) or placebo QD for 7 days. In the second cohort, participants were randomized 3:1 to receive TPIP 112.5 µg QD for 4 days followed by 225 µg QD for 3 days (titrated group) or placebo QD for 7 days (Fig. 1). Safety results from the two cohorts of participants in the single-dose phase were used to determine the titration step. Participants were admitted to the clinic for 10 days, during which daily doses of TPIP or placebo were administered for the first 7 days followed by 3 days of assessments and observations. Participants were discharged on day 10 and contacted via telephone for a safety follow-up approximately 30 days after the last dose.
The study was carried out in accordance with the ethical principles derived from international guidelines, including the Declaration of Helsinki and the Council for International Organizations of Medical Sciences International Ethical Guidelines, International Council for Harmonisation Good Clinical Practice Guideline, and applicable local regulatory requirements and laws. Before study entry, all participants provided written informed consent in compliance with US Title 21 Code of Federal Regulations, Part 50, which was submitted by the investigator to the institutional review board for the study site.
ParticipantsHealthy male and female nonsmokers aged 18 to 45 years who had a body mass index from 19.0 to 32.0 (calculated as weight in kilograms divided by height in meters squared) and no clinically significant abnormal findings from medical history, physical examination, vital sign measurements, 12-lead electrocardiogram, or clinical laboratory tests were eligible. Participants were excluded if they had a documented history of anaphylaxis or hypersensitivity to any drug including treprostinil and TPIP excipients, abnormal renal function (estimated glomerular filtration rate less than 60 mL/min/1.73 m2 by the Chronic Kidney Disease Epidemiology Collaboration equation), HIV infection, surgery requiring general anesthesia within 90 days, or malignancy within 5 years (excluding nonmelanoma skin cancer) before screening. Use of prescription medication (excluding hormonal birth control) or over-the-counter medications within 14 days of the first dosing day or throughout the study was not permitted. Concomitant medications deemed necessary for the welfare of the participant during the study were allowed at the investigator’s discretion. The complete list of inclusion and exclusion criteria are provided in Online Resource 1.
Study Drug TreatmentDosing of TPIP (7.5 mg dry powder per capsule packaged in a single actuation capsule containing 112.5 µg TP) or matching placebo occurred after an overnight fast for at least 10 h. Water was permitted except for 1 h before and 1 h after study drug administration when serial PK samples were collected. Study drug was administered via an actuated device held by study staff. Participants performed up to two inhalations per capsule over a period up to 15 min and remained seated or standing for at least 1 h after dosing. Active drug and placebo were identical in appearance, and all participants within their cohort were dosed with the same number of capsules. When multiple dose levels were required, the number of capsules administered was determined by the number of capsules required to achieve the highest desired dose level, and other dose levels were achieved by an appropriate mix of active and placebo capsules. An unblinded pharmacist dispensed the capsules in a manner consistent with maintaining the blinding.
Safety AnalysesSafety was assessed via monitoring and recording of AEs (including treatment-emergent AEs [TEAEs] and serious AEs), clinical laboratory test results (hematologic assessments, serum chemistry, and urinalysis), vital sign measurements (blood pressure, pulse rate, respiratory rate, and body temperature), and physical examination findings. Clinical laboratory testing occurred at screening, check-in, and day 4 in the single-dose phase and at day 8 and day 10 in the multiple-dose phase. On dosing days, vital signs were measured at screening, check-in, 15 min before dosing, and 0.5, 1, 2, 4, 8, and 12 h after dosing. Vital signs were also measured on nondosing days at times that coincided with those on dosing days (i.e., time-matched intervals at 4, 8, and 12 h after scheduled dosing).
AEs (defined as any untoward medical occurrence regardless of whether it is considered drug related) were coded by preferred term and system organ class using the Medical Dictionary for Regulatory Activities (MedDRA) v23.0. TEAEs were defined as any event that was not present before exposure to study drug or any event that worsened in intensity or frequency after exposure. AEs were assessed from administration of the first dose until participants were discharged from the clinic and up to approximately 30 days after the last dose. All AEs were followed up until they were resolved, stable, or judged clinically insignificant by the investigator.
PK AnalysesPK samples were analyzed by a validated liquid chromatography coupled with tandem mass spectrometry assay for treprostinil and TP in plasma samples with potassium EDTA as coagulant. The treprostinil analysis methods consisted of a BetaSil C8 Column (5 µm; ThermoFisher Scientific), [13C2D1]treprostinil as an internal standard, and a mobile-phase gradient of 5 mM ammonium bicarbonate and methanol. Treprostinil and its internal standard were detected under a negative ionization mode at m/z 389.2 → 331.1 and 392.1 → 332.1, respectively. The TP analysis method consisted of a C18 column (Poroshell 120 EC-C18 30 × 3.0 mm, 2.7 μm [Agilent]), TP-d4 as an internal standard, and a mobile-phase gradient of 0.1% trifluoroacetic acid in 10 mM ammonium acetate and acetonitrile. TP and its internal standard were detected under electrospray-positive ionization at m/z 632.5 → 355.3 and 636.5 → 355.3, respectively. The quantitation limits for treprostinil and TP were 10 pg/mL and 100 pg/mL, respectively.
In the single-dose phase, serial blood samples were collected on day 1 before dosing and 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24, 36, 48, and 72 h after dosing. In the multiple-dose phase, serial blood samples were collected on day 1 before dosing and 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, and 12 h after dosing; before dosing only on days 2, 3, 4, 5, and 6; and before dosing on day 7 and 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24, 48, and 72 h after dosing.
PK parameters derived from plasma samples were calculated using a noncompartmental analysis with Phoenix WinNonlin version 8.0 or higher (Certara, Inc) or SAS version 9.4 or higher (SAS Institute Inc), including the time to maximum plasma concentration (Tmax), Cmax, AUC from time 0 to the last time with quantifiable concentration (AUClast), AUC from time 0 to 24 h (AUCτ), AUC from time 0 to infinity (AUC0−∞), t1/2, total drug clearance following extravascular administration (CL/F), and volume of distribution at the terminal phase (Vd/F).
Statistical AnalysesPlasma concentrations below the limit of quantification (BLQ) were treated as zero for descriptive statistics. For PK parameter calculations, predose data on day 1 were treated as zero. All postdose BLQ values were considered missing.
Dose proportionality was assessed in the single-dose phase using the power regression model for AUCs and Cmax defined by ln[PK parameter] = β0 + β1ln[dose]. The null hypothesis was that AUCs and Cmax were dose proportional, or slope (β1) was equal to 1. Dose proportionality was concluded if the 90% confidence interval (CI) of the slope β1 was entirely within [1 + ln(0.5)/ln(r), 1 + ln(2)/ln(r)], where r was a ratio of highest dose to lowest dose that described the dose range. Steady state was tested in the multiple-dose phase by visual inspection of individual plasma trough levels from days 1 to 7.
The safety population consisted of participants who received at least one dose of TPIP or placebo. The PK population consisted of participants who received at least one dose of TPIP and had at least one measurable postdose plasma concentration. Sample sizes were not based on a formal statistical power calculation but were chosen to obtain reasonable evidence of safety and tolerability without exposing an undue number of participants to TP during this phase 1 study. The sample size was based on previous experience in phase 1 studies and not a formal statistical power calculation. All analyses were performed using SAS version 9.4 or higher (SAS Institute Inc).
留言 (0)