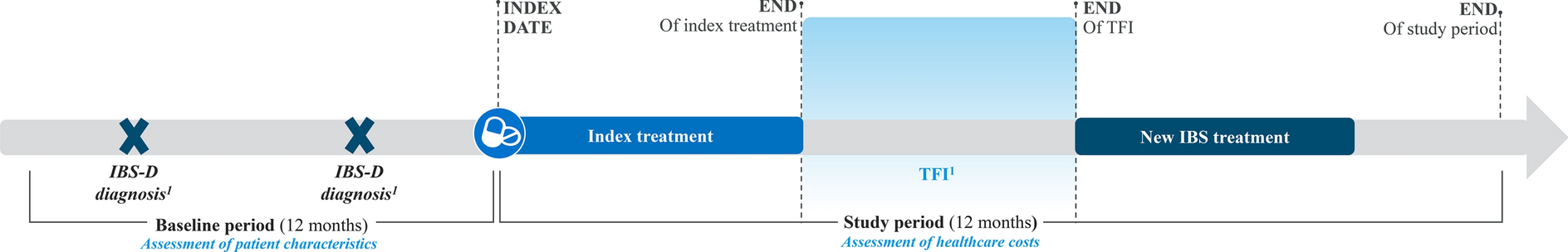
Study Design
The EX-DKD study was a multicenter (22 sites), open-label, prospective study conducted in Japan from December 2019 to May 2022 (Fig. S1 in the supplementary material), and participating institutions and representative physicians are listed in Table S1 in the supplementary material.
Compliance with Ethics Guidelines
The study protocol was approved by the Okayama University Certified Review Board (CRB6180001) and prospectively registered with the Japan Registry of Clinical Trials (jRCTs061190027; https://jrct.niph.go.jp/en-latest-detail/jRCTs061190027). The study was conducted in accordance with the principles of the Declaration of Helsinki 1964, and its later amendments, and the Clinical Trials Act in Japan. All patients provided written informed consent before enrollment.
Patients
The inclusion criteria were as follows: patients aged 20 to < 85 years, with a diagnosis of type 2 DM (glycated hemoglobin < 9%), for whom home BP measurements could be obtained, who had received prior RAS inhibitor or RAS inhibitor plus CCB treatment before obtaining informed consent, had sitting office SBP of 130 to < 180 mmHg and/or DBP of 80 to < 110 mmHg at the start and end of the pre-study observation run-in period, had a UACR of < 1000 mg/gCr during the pre-study observational run-in period, and had a creatinine-based estimate of the glomerular filtration rate (eGFRcreat) of 30 to < 60 mL/min/1.73 m2. Major exclusion criteria were as follows: patients with secondary or malignant hypertension, type 1 DM, non-diabetic chronic kidney disease with increased or decreased doses of steroids or immunosuppressive drugs within 3 months before obtaining informed consent or with plans to increase or decrease doses of these drugs within the following 4 months, nephrotic syndrome, acute glomerulonephritis, complication or history of orthostatic hypotension, rapidly progressive glomerulonephritis, ankle-brachial index ≤ 0.9 in patients with symptoms caused by arteriosclerosis obliterans, cerebrocardiovascular disease, severe hepatic dysfunction, and serum potassium level > 5.0 mEq/L at the end of the pre-study observation run-in period. On the basis of baseline UACR, all patients were divided into two albuminuria subcohorts or three UACR subcohorts as follows: patients with a UACR of < 30 mg/gCr and 30 to < 1000 mg/gCr were assigned to the no albuminuria and albuminuria subcohorts, respectively, and patients with a UACR of < 30 mg/gCr, 30 to < 300 mg/gCr, and 300 to < 1000 mg/gCr were assigned to the A1, A2, and A3 subcohorts, respectively.
Treatments
After a 4-week run-in period, esaxerenone was initiated at a dose of 1.25 mg/day, and then could be gradually increased to 2.5 mg/day (week 4) and to 5 mg/day (week 8) on the basis of BP and serum potassium level monitoring. The entire treatment period was 12 weeks. Prior treatment with RAS inhibitor (or RAS inhibitor plus CCB) was continued at a constant dose throughout the study, and the dose and type of antihypertensive and antihyperglycemic drugs could not be changed. The following concomitant drugs were prohibited from 4 weeks before the treatment start to the end of the treatment (EOT) period or the time of discontinuation: antihypertensive and antianginal drugs (angiotensin receptor blockers [ARBs], angiotensin-converting enzyme inhibitors, CCBs, renin inhibitors, α-blockers, β-blockers, αβ-blockers, other sympatholytic agents, vasodilators), diuretics (thiazide, thiazide-like, loop, potassium-sparing diuretics), aldosterone antagonists, potassium preparations, non-steroidal anti-inflammatory drugs, and calcium polystyrene sulfonate.
Measurement of BP
The office BP and pulse rate were measured twice at each visit (at baseline; 2, 4, 8, and 12 weeks; end of the study; and discontinuation of the study), and the average of the two measurements was used. Office BP was measured at intervals of at least 3 h after meals and after at least 5 min of rest in a sitting position. When the clinical blood sampling was carried out, the BP and pulse rate were measured prior to the blood sampling. Home BP was self-measured twice (at morning and bedtime) using an upper arm cuff sphygmomanometer within the last 5 days of the patient’s visit, and the average of the two measurements was used. The sphygmomanometer owned by the patients was used throughout the study period. Morning home BP was measured after urination within 1 h after waking up and before breakfast, medication, and caffeine intake. Bedtime home BP was measured over 1 h after bathing, drinking, or caffeine intake before bedtime.
Measurement of Other Outcomes
At each visit (at baseline; 4, 8, and 12 weeks; and the time of discontinuation), the albumin and creatinine concentrations in the collected spot urine samples were measured by a central measurement laboratory (SRL, Inc., Tokyo, Japan), and the UACR was calculated using the following formula: UACR (mg/gCr) = urinary albumin (µg/mL)/urinary creatinine (mg/dL) × 100. Plasma aldosterone concentration (PAC) and plasma renin activity (PRA) were measured at baseline, 12 weeks, and at discontinuation in the central measurement laboratory (SRL, Inc.). PAC and PRA were measured after resting in the supine position for at least 30 min. Urinary concentrations of sodium, potassium, creatinine, protein/creatinine ratio, and biomarkers including liver-type fatty acid binding protein, N-acetyl-β-d-glucosaminidase, β2-microglobulin, angiotensinogen, and 8-hydroxydeoxyguanosine were measured at baseline, 12 weeks, and at discontinuation in the central measurement laboratory (SRL, Inc.). The eGFRcreat was calculated as follows: 194 × serum creatinine−1.094 × age−0.287, multiplied by 0.739 for female patients [24].
Efficacy Endpoints
The primary efficacy endpoint was the change in morning home SBP and DBP from baseline to EOT. Secondary efficacy endpoints were the change in bedtime home and office SBP and DBP from baseline to EOT, the time course change of home (morning, bedtime) and office SBP and DBP during the study, the achievement rate of target BP levels (SBP/DBP, home < 125/75 mmHg and office < 130/80 mmHg) at 12 weeks [11], the change and percentage change in UACR from baseline to EOT, and changes in serum and urinary biomarkers from baseline to 12 weeks.
Safety Endpoints
The safety endpoints included treatment-emergent adverse events (TEAEs), laboratory tests, vital signs, and 12-lead electrocardiography. The incidence of serum potassium level ≥ 5.5 and ≥ 6.0 mEq/L, time course changes in eGFR, and change from baseline in eGFR were also assessed as safety endpoints.
Statistical Analysis
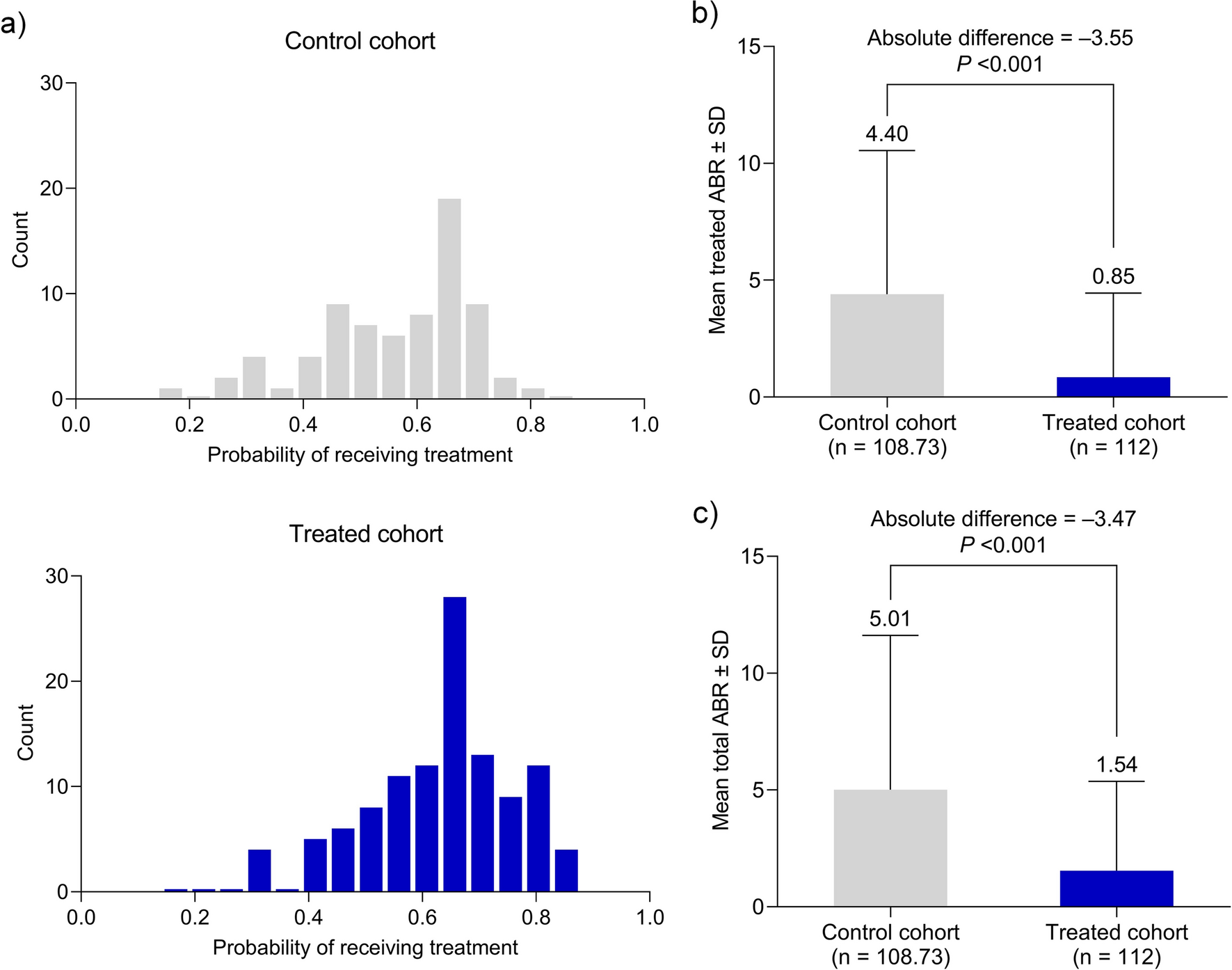
We assumed that the change in sitting BP ± SD with esaxerenone would be − 8.7 ± 19.0 mmHg for SBP and − 4.9 ± 11.0 mmHg for DBP based on previous studies [25, 26]. Under this assumption, the statistical power was calculated as more than 90% when the analysis population was 55, and the significance level was set at a two-sided p value < 0.05. Considering five patients excluded from the analyses, the target sample size was set at 60 patients in each subcohort (no albuminuria and albuminuria subcohorts). Efficacy endpoints were analyzed in the full analysis set (FAS) that included all patients who provided informed consent, met the eligibility criteria, took at least one dose of esaxerenone, and had at least one efficacy measurement recorded. Point estimates and 95% CIs for the difference between baseline and EOT values were calculated and compared using the paired t test. EOT values were calculated by taking the average of measurements at the last two visits in the treatment period. The missing values at the EOT for sitting BP and UACR were imputed by the last observation carried forward method. Missing PAC, PRA, urinary biomarkers, and safety endpoints were not imputed. Safety endpoints were evaluated in the safety analysis set, defined as all enrolled patients who took at least one dose of esaxerenone, and were summarized using descriptive statistics. TEAEs were coded by System Organ Class and Preferred Term according to the Medical Dictionary for Regulatory Activities (MedDRA), version 24.1. Subgroup analysis was conducted according to baseline UACR (A1, A2, and A3). Multiplicity was not adjusted because of the small sample size, as this was an exploratory study. The significance level was set as 5% (two-sided). All statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).



留言 (0)