
記住我



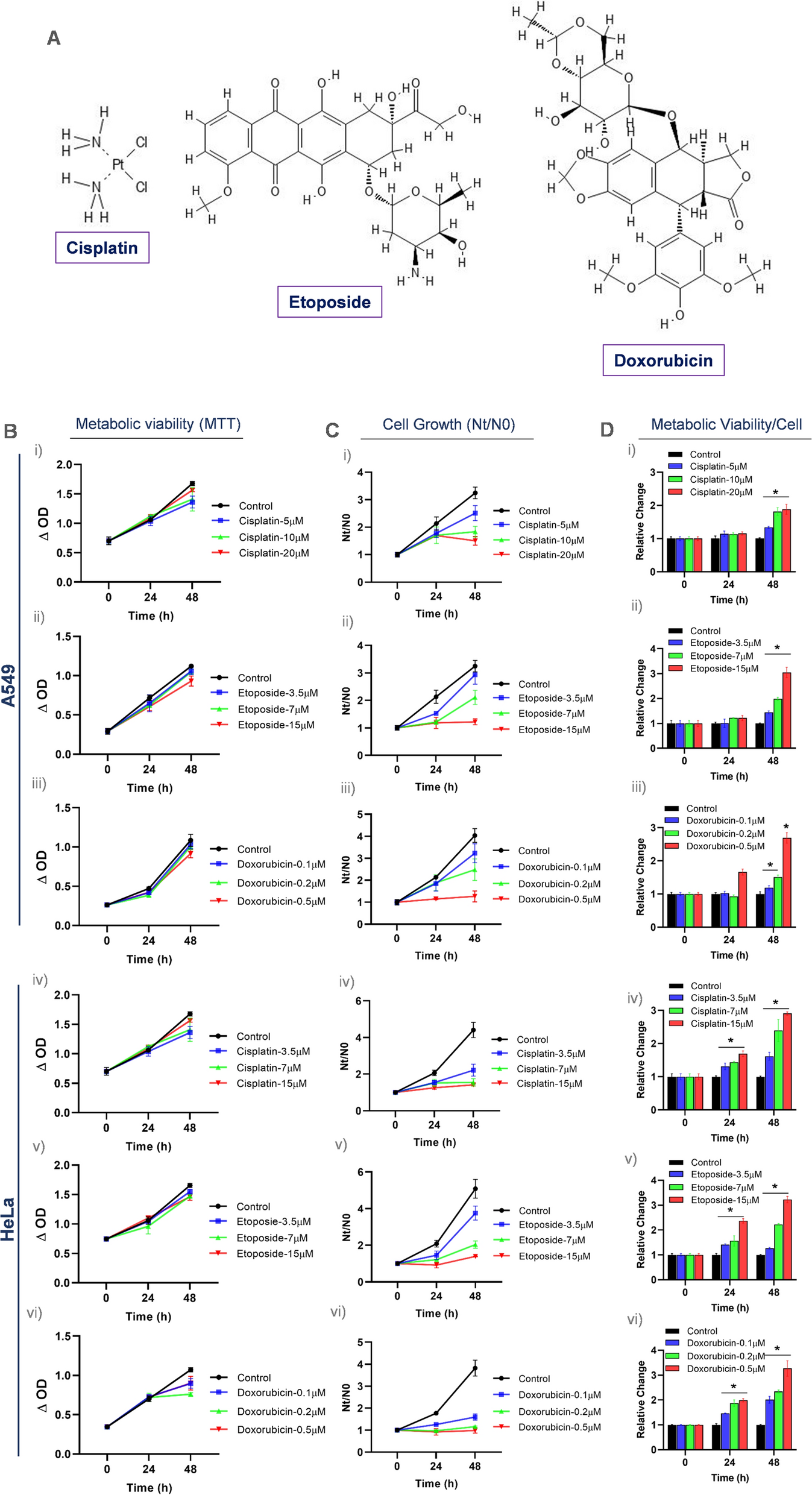
Cell culture media used in culture procedures (Table 1) were modifications of previously described media for mouse tracheal cell culture (You et al. 2002) as well as for ALI culture of murine and porcine OEC (Chen et al. 2017). The cell culture medium DMEM/Ham’s F12 (1:1, FG 415), FBS (S 0115, Lot 1030B) and amphotericin B were obtained from Merck Millipore (Darmstadt, Germany), while other chemicals, media and supplements were purchased from Sigma-Aldrich (Taufkirchen, Germany) unless otherwise stated.
Table 1 Media compositions used for compartmentalized cell culture AnimalsOviduct tissue samples of 33 domestic cats (F. catus) were obtained from the Animal Rescue Shelter or Veterinarians in Berlin, Germany. They were collected by the local veterinarians after routine gonadectomy. The experimental protocols were approved by the Ethics Committee of the Leibniz Institute for Zoo and Wildlife Research (2013-05-05).
Tissue and sample preparationOvaries with oviducts were transported to the laboratory in transport medium at 4 °C and were processed within 5 h. However, the storage time between gonadectomy and transport to the laboratory varied between 1 and 6 h. The organs markedly differed in size and ovarian status. Oviducts from both, late follicular phase ovaries (follicles larger than 2 mm in diameter visible) and inactive ovaries (neither follicles nor corpora lutea visible) were used.
Isolation of FOEC was conducted according to a previously published protocol (Eder et al. 2020). Briefly, after removal of the ovary and surrounding tissues, oviducts were washed in washing medium. The oviducts were injected with 1 mg/ml collagenase 1 A (C5894) in PBS (D8537) and incubated for 30 min at 38 °C. Epithelial cells from isthmus and ampulla were squeezed out separately with the outer edge of a scissor onto a glass slide, flushed into 600 µl FOEC-BASIC in a 4-well dish and pre-cultured for 16 h at 38.5 °C, 5% CO2. If both oviducts of one cat were available, the two samples of each region were pooled.
During the pre-culture, extracted cell associations formed three-dimensional vesicles. After pre-culture, FOEC-vesicles mainly consisting of viable cells were separated from vesicles comprising many dead cells (Eder et al. 2020). They were resuspended in 10 × trypsin/EDTA (59418 C) solution for subsequent single cell isolation. After 10 min digestion at 38 °C in a water bath, the reaction was stopped by adding 750 µl FBS. Single cell isolation was completed by gentle pipetting through a wide pipet tip and filtering through a cell strainer of 40 μm pore size. Cell concentrations were determined in a counting chamber. FOEC suspensions were centrifuged for 5 min at 200 × g, pellets were resuspended with proliferation medium (FOEC-PROL) and adjusted to a cell concentration of 1 × 106/ml.
Long-term culture of FOECFor the long-term culture, 24-well hanging inserts (PET, pore size 0.4 μm, non-transparent, Millipore, Switzerland) were used. Inserts were coated with human placenta collagen (C5533) according to the manufacturer’s instructions.
Depending on the number of isolated viable cells, 1–2 × 105 FOEC were seeded onto the apical side of each insert. Each well (basal side of insert) was filled with 1 ml FOEC-PROL. All samples were maintained submerged during the proliferation phase for 3 to 5 days. Once a confluent cell monolayer was established, cells were either grown at the ALI with 1 ml FOEC-DIFF in the basolateral compartment and without addition of medium on the apical side, or with 1 ml FOEC-DIFF in the basolateral and 50 µl FOEC-DIFF in the apical compartment of the inserts (LLI). Medium in the basolateral compartment (ALI approach) or in both the basolateral and apical compartment (LLI approach) was changed twice a week. During a culture period of 3 weeks the FOEC were incubated in humidified atmosphere with 5% CO2 at 38.5 °C.
Histology and morphological evaluationHistological processing was performed as previously described by our group (Chen et al. 2013a, b). Briefly, the cell layer together with the filter support was fixed in Bouin`s solution, stabilized in agarose and post-fixed in 4% phosphate buffered formaldehyde. After dehydration in an ascending ethanol series, the samples were embedded in paraffin. Samples were cut into 3 μm sections, stained with haematoxylin/eosin (HE) and microscopically evaluated for epithelial differentiation using the criteria depicted in Fig. 1.
Fig. 1
Criteria applied for the morphological evaluation of FOEC after 3 weeks of culture (OE oviduct epithelium)
ImmunohistochemistryImmunolocalization of marker proteins for cilia development (acetylated tubulin) and oviduct specific secretory activity (oviduct-specific glycoprotein) was conducted in morphologically differentiated cultures. Antigen retrieval was performed either enzymatically (acetylated tubulin) by incubation with 0.06% trypsin, pH 7.8 or via heat-induced antigen retrieval (oviduct-specific glycoprotein) using sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0). Unspecific binding sites were blocked with 5% BSA and 10% goat serum in PBS (30 min, room temperature). Slides were incubated with the primary antibodies mouse monoclonal anti-human acetylated tubulin (Sigma T7451; 1:1000 in PBS with 1% BSA) or rabbit polyclonal anti-human oviduct-specific glycoprotein (Abcam ab118590; 1:500 in PBS with 1% BSA), respectively (overnight, 4 °C). The corresponding secondary antibodies were goat anti-mouse IgG, Alexa 568 (Invitrogen A-11031; 1:40 in PBS with 1% BSA) and goat anti-rabbit IgG, Alexa 647 (Invitrogen A-21245; 1:200 in PBS with 1% BSA), respectively, and were applied for 1 h at room temperature. Negative controls were performed by omitting the primary antibody. SYBR Green I (Mobitec, Berkheim) was used for nuclei counterstaining. Pictures were captured using a Zeiss LSM800 equipped with fluorescence optics and ZEN software.
留言 (0)