
記住我

The kidneys are greatly affected by hemodynamics under heart failure in a phenomenon called cardiorenal syndrome [1,2]. Renal venous congestion by abdominal venous stasis is deeply associated with cardiorenal syndrome and relates to the progression of heart failure [3–5]. Renal interstitial hydrostatic pressure (RIHP) is regulated by renal venous pressure (RVP) [6,7]. Several animal studies have shown that acute RVP elevation increases RIHP and decreases renal blood flow [8,9]. Ligation of the inferior vena cava (IVC) above the renal veins also raises RVP [10] and induces chronic renal dysfunction [11,12], especially renal fibrosis with excess extracellular matrix in the renal interstitium [13].
We have recently developed a novel rat renal venous congestion model with RHIP elevation, tubular dilatation, and interstitial fibrosis [14]. And the reduction of RIHP with renal capsulotomy ameliorated this renal fibrosis. Especially in the outer medulla, transgelin (Tagln) and platelet-derived growth factor receptors (PDGFRs) were upregulated. Pericyte detachment was also observed around the expanded vasa recta, which leads to high circumferential (hoop or tangential) stress, in the congested kidneys.
Renal pericytes stabilize vascular networks through the receptors of angiogenic growth factors, including PDGFRs [15,16]. Pericytes also contribute to the renal myofibroblast pool in renal fibrogenesis via pericyte-myofibroblast transition (PMT) [17–20], and their detachment from capillaries after acute/chronic kidney injury may be critical in driving chronic kidney disease progression [21]. Activation of the PDGFR pathway is a trigger for PMT in progressive kidney disease [22]. Thus, PMT blockage by PDGFR inhibitors could be a novel therapeutic target against tubulointerstitial fibrosis. Indeed, imatinib, a potent inhibitor of multiple receptor tyrosine kinases, has been used as an inhibitor of PDGFRs, resulting in renoprotective effects in several animal experiments [22–33].
We, therefore, hypothesized that the activation of the PDGFR pathway is responsible for renal fibrosis in renal congestion. The renoprotective effect of imatinib against renal injury was investigated in our rat renal congestion model. The anti-fibrotic effect of the PDGFR inhibition in pericytes was verified in vitro cell culture using imatinib and crenolanib, a highly selective PDGFR inhibitor [34,35].
METHODS AnimalsAll animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Tohoku Medical and Pharmaceutical University Animal Experiment Committee (registration number: A18019-a, A19039-cn, A20005-cn, and A21008-cn). Male Sprague-Dawley rats (6 weeks old, 180–220 g; Japan SLC, Shizuoka, Japan) were housed in environmentally controlled rooms under a 12-h light/dark cycle, and fed pellets (CE-2; CLEA Japan, Tokyo, Japan) and water ad libitum. Animal experiments were performed after at least 5 days of acclimatization, and were designed to use all animals for the analysis except for those that died or had animal welfare issues during the experiments.
Inferior vena cava ligation and imatinib administrationIVC ligation was performed according to our previous study [14] with slight modification. Briefly, rats were anesthetized with a mixture of medetomidine (0.15 mg/kg body weight; Maruishi Pharmaceutical, Osaka, Japan), midazolam (2.0 mg/kg body weight; Astellas Pharma, Tokyo, Japan), and butorphanol (2.5 mg/kg body weight; Meiji Seika Pharma, Tokyo, Japan). After midline abdominal incision, the IVC between the renal veins was ligated on a temperature-controlled (38°C) surgical table (Figure S1A, Supplemental Digital Content, https://links.lww.com/HJH/B967). After abdominal closure, penicillin (300 000 U/kg body weight) and buprenorphine (0.05 mg/kg body weight) were administered intramuscularly. Three days after surgery, under anesthesia, blood and urine were collected from the abdominal aorta and urinary bladder, respectively, and the rats were euthanized under anesthesia. The kidneys, heart, and liver were removed, and immediately weighed and sectioned. The tissues for histological analysis were fixed with 10% formalin (Mildform; Wako Pure Chemical Industries, Osaka, Japan) and embedded in paraffin. Both kidneys were divided into the cortex and outer medulla, and were kept in RNA Later (Invitrogen, Carlsbad, California, USA) for RNA or snap-frozen in liquid nitrogen for protein analyses. Biochemical examinations were performed by Nagahama Life Science Laboratory (Nagahama, Japan).
Rats were randomly selected and injected intraperitoneally with saline (saline group, n = 12; Fuso Pharmaceutical Industries, Osaka, Japan) or imatinib mesylate (imatinib group, n = 12, 20 mg/kg, dissolved in saline; Tokyo Chemical Industry, Tokyo, Japan) from the day before the surgery up to two days later (Figure S1A, Supplemental Digital Content, https://links.lww.com/HJH/B967). The dose of imatinib and sample size were determined based on previous reports [22–25] and our preliminary experiments.
Physiological analysisIVC pressure was measured using the PowerLab system (AD Instruments, Sydney, Australia), as described in our previous report [14]. Briefly, rats were anesthetized with ketamine (20 mg/kg body weight, intraperitoneally; Toho Pharmaceutical, Tokyo, Japan) and thiobutabarbital (50 mg/kg body weight, intramuscularly; Sigma-Aldrich, St. Louis, Missouri, USA) on a temperature-controlled (37°C) surgical table, and respiration was maintained by inserting a PE 240 catheter into the trachea. Upstream (junction of the left renal vein and the vena cava) and downstream (junction of the right renal vein and the vena cava) IVC pressures during IVC ligation were measured by PE 50 catheters inserted from the bilateral femoral veins and recorded for a total of 60 min (30 min before and 30 min after IVC ligation) after at least 30 min for equilibration with the PowerLab system (Figure S1B, Supplemental Digital Content, https://links.lww.com/HJH/B967). Upstream IVC pressure on day 3 after IVC ligation was measured by a PE 50 catheter inserted from the left femoral vein and recorded for 30 min after at least 30 min for equilibration with the PowerLab system (Figure S1C, Supplemental Digital Content, https://links.lww.com/HJH/B967). Saline containing 2.0% bovine serum albumin (BSA) was infused through the cervical vein catheter at 1.0 ml/100 g body weight/h during the IVC pressure measurements.
Ex vivo micro-computed tomography scanningFor the renal vascular structure visualization, Microfil-perfused kidneys were imaged by micro-computed tomography (μCT; CosmoScan GX II; Rigaku, Tokyo, Japan). The kidneys were perfused with ice-cold phosphate-buffered saline and 4% paraformaldehyde (PFA) under anesthesia. This was followed by infusion with 10 ml of the radiopaque contrast agent Microfil MV-112 (5:4 ratio of MV-diluent: MV-compound and 5% MV-curving agent [36]; Flow Tech, Carver, Massachusetts, USA). After 2–3 h of Microfil solidification, the kidneys were removed and incubated in 4% PFA at 4°C overnight. The kidneys were then scanned by μCT under the following conditions: X-ray voltage, 90 kV; current, 88 μA; field of view (FOV), 25 mm; voxel size, 50.0 μm x 50.0 μm × 50.0 μm; and scan time, 4 min (high resolution).
Cell cultureHuman pericytes from the placenta (hPC-PL; Promo Cell GmbH, Heidelberg, Germany) were cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Waltham, Massachusetts, USA)/20% fetal bovine serum (FBS; Life Technologies, Carlsbad, California, USA) at 37°C under 5% CO2. Cells were seeded at 5 × 105 cells/well in 12-well plate (Thermo Fisher Scientific), serum-starved in DMEM/1% FBS overnight, and then incubated for 24 h in following conditions: vehicle, 1.0 μg/ml transforming growth factor-β1 (TGFB1; R&D Systems, Minneapolis, Minnesota, USA), 1.0 μg/ml TGFB1 and 25 μmol/l imatinib, and 1.0 μg/ml TGFB1 and 1.0 μmol/l crenolanib (Selleck, Houston, Texas, USA) in DMEM/20% FBS. The concentration of the reagents was determined based on a previous report [34].
Quantification of RNA expression levelsTotal RNA was extracted from the rat kidneys using ISOGEN (NIPPON GENE, Tokyo, Japan), and from the cultured cells using a silica-based spin column (FastGene RNA Basic Kit; Nippon Genetics, Tokyo, Japan). cDNA was synthesized from the total RNA using random hexamer (Invitrogen) and SuperScript III First-Strand reverse transcriptase (Invitrogen). The target cDNAs were amplified in duplicate with specific primers (Table S1, Supplemental Digital Content, https://links.lww.com/HJH/B967) using THUNDERBIRD SYBR qPCR Mix (Toyobo, Osaka, Japan) and CFX Connect (Bio-Rad, Hercules, California, USA). The relative mRNA expression levels were normalized to the values of ribosomal protein lateral stalk subunit P2 (Rplp2) in rat kidneys, or actin beta (ACTB) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in human cells.
Imatinib-binding genes were selected according to the previous report [37]. The microarray data set (GEO accession number: GSE114031) that we compared the right control kidney and the left congested kidney in our previous study [14] was also used to check the mRNA expression levels of the imatinib-binding genes.
Western blottingKidneys were homogenized in lysis buffer (9803; Cell Signaling Technology, Danvers, MA) containing 1.0 mmol/l phenylmethylsulfonyl fluoride (Thermo Fisher Scientific) and protease inhibitor cocktail (Roche, Basel, Switzerland). Twenty microgram of proteins mixed with Laemmli sample buffer (Bio-Rad) and 2.5% mercaptoethanol were separated by 4–20% Mini-PROTEAN TGX Gels (Bio-Rad) and transferred onto trans-blot turbo transfer pack membranes (Bio-Rad). After incubation with a blocking reagent (PVDF Blocking Reagent for Can Get Signal; Toyobo), the membranes were incubated with antigen-specific antibodies (Table S2, Supplemental Digital Content, https://links.lww.com/HJH/B967) overnight at 4°C. Immunoreactive signals were visualized using a horseradish peroxidase-conjugated secondary antibody (1:5000; Santa-Cruz Biotechnology, Dallas, Texas, USA), an enhanced chemiluminescence system (Thermo Fisher Scientific), and Amersham Imager 600 (GE Healthcare, Buckinghamshire, UK). The relative expression level of each protein was normalized to GAPDH.
Histological analysisFour-micrometer-thick sections were deparaffinized with xylene and hydrated in gradient ethanol and distilled water. The sections were stained with hematoxylin-eosin and Elastica-Masson for routine histological analysis in the Technical Service Division of Tohoku Medical and Pharmaceutical University. The Picrosirius Red staining (Polysciences, Warrington, Pennsylvania, USA) was performed according to the manufacturer's instruction.
For immunohistochemistry, after deparaffinization, the antigens were retrieved in an autoclave for 5 min at 121°C in 10 mmol/l citrate buffer (pH6.0) or 1.0 mmol/l ethylenediaminetetraacetic acid buffer (pH 9.0). Then, the antigens were reacted with antigen-specific antibodies (Table S2, Supplemental Digital Content, https://links.lww.com/HJH/B967) overnight at 4°C. On the next day, the sections were incubated with biotinylated secondary antibodies (Abcam, Cambridge, UK), and the reaction was visualized by streptavidin-conjugated horseradish peroxidase (SouthernBiotech, Birmingham, Alabama, USA) and 3,3′-diaminobenzidine (DAB; Dojindo, Kumamoto, Japan), or fluorophores (Alexa 488 or Alexa 555; Molecular probes, Carlsbad, California, USA). The nuclei were counterstained with hematoxylin or Hoechst 33342 (Molecular Probes). The slides were digitized by a microscope (BZ-X 710; KEYENCE, Tokyo, Japan) or confocal microscopy (Leica TCS SP8; Leica Microsystems, Wetzlar, Germany).
For double-labeling immunofluorescence using primary antibodies with the same host species, the tyramide signal amplification (TSA)-based multiplex immunofluorescence method was used [38]. The sections were deparaffinized, retrieved antigens, and incubated with the first rabbit primary antibody (1:500, anti-NG2, AB5320; Millipore, Temecula, California, USA), biotinylated secondary antibody, and streptavidin-conjugated horseradish peroxidase as described above. Then, according to the manufacturer's protocol, covalent deposition of fluorophores around the recognition point of the primary antibody was developed with the catalytic activity of horseradish peroxidase and iFluore 488 Styramide reagent (45020; AAT Bioquest, Sunnyvale, California, USA). After the primary and secondary antibodies were stripped from the sections by microwave for 10 min in 10 mmol/l citrate buffer, the sections were reacted with the subsequent rabbit primary antibody (1:4000, anti-CD11B, ab133357; Abcam or 1:200, anti-CD206, 24595S; Cell Signaling) and fluorophore-conjugated secondary antibody (1:1000, A21430; Life Technologies), counterstained with Hoechst 33342, and mounted with ProLong Gold Antifade mounting medium (Molecular Probes).
To measure collagen positive area around the vasa recta, the area of the Picrosirius Red staining was calculated digitally using Hybrid Cell Count BZ-H3C application (Figure S2, Supplemental Digital Content; KEYENCE, https://links.lww.com/HJH/B967) following the previous report [39].
Low-vacuum scanning electron microscopyThe ultra-structure of the vasa recta was observed by low-vacuum scanning electron microscopy (LV-SEM, Miniscope TM4000; Hitachi High-Technologies, Tokyo, Japan), as previously described [14]. Briefly, 4-μm-thick sections stained with Pt-blue solution (TI-blue small kit; Nisshin EM, Tokyo, Japan) were captured at an acceleration voltage of 15 kV and 30 Pa. Four fields of the vasa recta were randomly selected from each slide, and the diameter of the vasa recta was manually measured by ImageJ software.
Statistical analysisData collection, interpretation, and statistical analysis were conducted by separate researchers, who were blinded to the procedures. Continuous values are presented as the means ± standard error of the means (SEM). Statistical comparisons were conducted using Mann−Whitney U test for two-group comparisons, and Kruskal−Wallis test followed by Steel−Dwass test for multiple comparisons. P values <0.05 were considered significant.
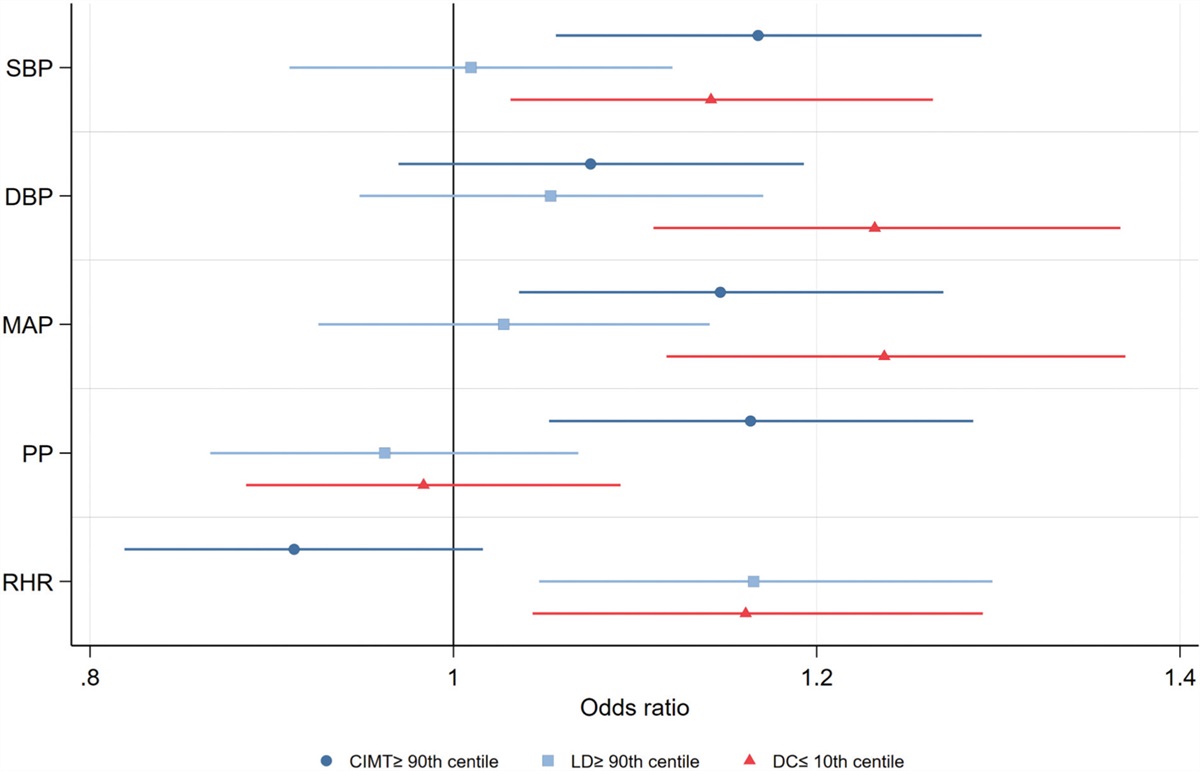
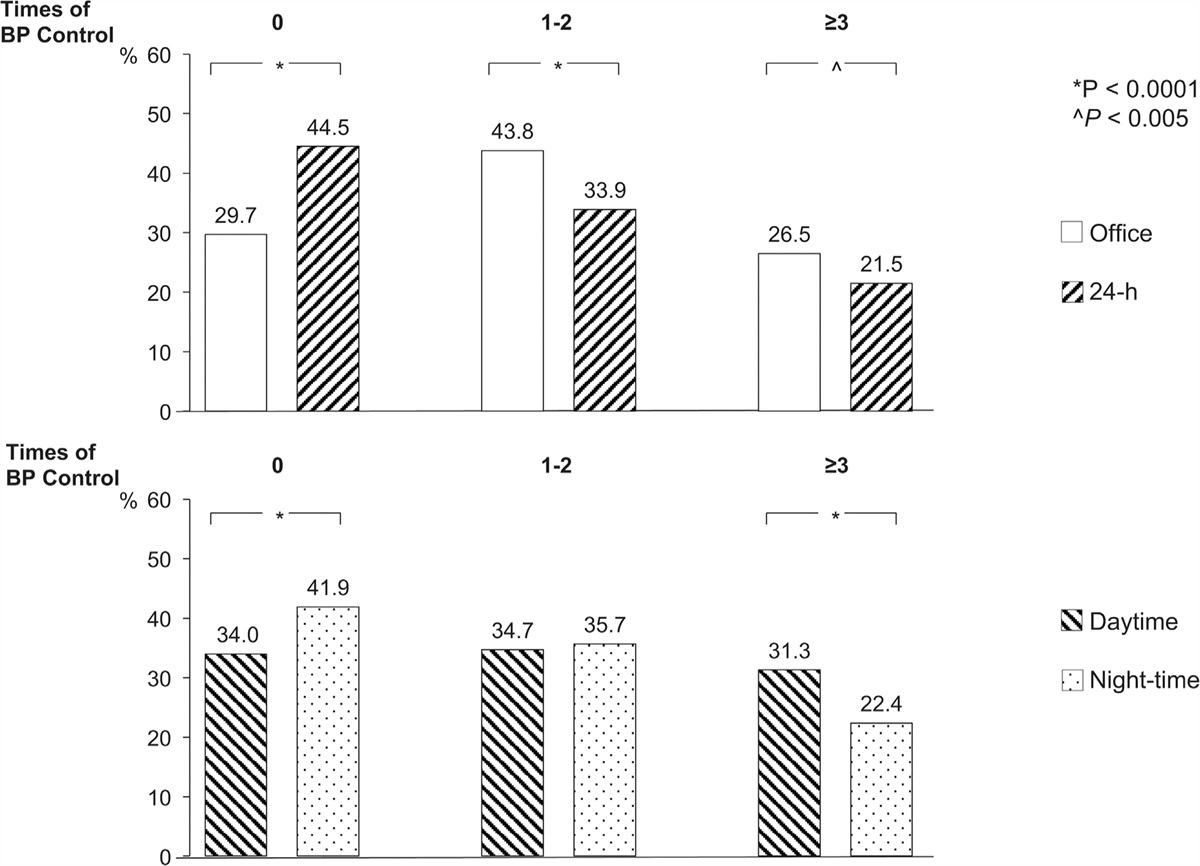
RESULTS Changes in morphology and biochemical parametersNo anatomical vein abnormalities were observed, and no rats died during the experimental period. Consistent with the previous report [14], the upstream IVC pressure immediately increased to around 20 mmHg (saline: 18.8 ± 0.5; and imatinib: 18.3 ± 0.8 mmHg) after IVC ligation, and kept high at around 10 mmHg (saline: 10.3 ± 1.0; and imatinib: 10.1 ± 0.7 mmHg) on the day 3 after IVC ligation in both the imatinib and saline groups (Fig. 1a and b). Imatinib had no effect on IVC pressure elevation in either the acute or sub-acute phase after IVC ligation. The weight of the left congested kidney was significantly elevated compared to that of the right control kidney (Fig. 1c). Imatinib significantly attenuated the elevated weight of the congested kidney. In contrast to kidney weight, liver and heart weight were not affected by imatinib (Fig. 1d and e).
 FIGURE 1:
FIGURE 1: Effect of imatinib on inferior vena cava (IVC) pressure and organ weight. (a) Upstream (junction of the left renal vein and the vena cava) and downstream (junction of the right renal vein and the vena cava) IVC pressures during IVC ligation in the imatinib-treated (n = 5) and saline-treated (n = 5) group. After at least 30 min for equilibration and 30 min IVC pressure measurement as “Baseline”, IVC between the renal veins was completely ligated with downstream catheter and IVC pressures were measured for 30 min as “After IVC ligation”. ∗P < 0.05 versus baseline; ns: P > 0.05 by Mann−Whitney U test. (b) Upstream IVC pressure on day 3 after IVC ligation in the imatinib-treated (n = 6) and saline-treated (n = 6) group. (c−e) Alteration of kidney (c), liver (d), and heart (e) weights in the imatinib-treated (n = 12) and saline-treated (n = 12) group. The weight of each organ was normalized to body weight. Data are presented as individual values and mean±SEM (b−e). ∗∗P < 0.01; ∗∗∗P < 0.001; ns: P > 0.05 by Steel−Dwass test.
Although the morphology of the pericytes around vasa recta was hardly visible by double-labeling immunofluorescence staining of pericyte marker (NG2) and vascular endothelial marker (PECAM1) (Figure S3, Supplemental Digital Content, https://links.lww.com/HJH/B967), vasa recta expansion and pericyte detachment, triggers in our congestion model [14], were detected on μCT and LV-SEM images and were not different between imatinib- and saline-treated congested kidneys (Fig. 2a and b). The injury stimulated phosphorylation/activation of PDGFRB around the vasa recta in the congested kidney was suppressed by imatinib (Fig. 2c and d).
 FIGURE 2:
FIGURE 2: Effect of imatinib on pericyte detachment and PDGFR activation. (a) Representative high-resolution ex vivo μCT images of the kidneys of imatinib- and saline-treated rats after Microfil perfusion. Left panels: a midline sagittal cut. Right panels: a superficial frontal cut from the arrow side. (b) Top: representative images of pericytes (red arrowhead) in the descending vasa recta (VR) by low-vacuum scanning electron microscopy. Scale bar = 20 μm. Bottom: quantification of VR diameter. n = 80 (saline-treated right control kidney), 105 (saline-treated left congested kidney), 96 (imatinib-treated right control kidney), and 99 (imatinib-treated left congested kidney). (c) Immunoblots of PDGFRB phosphorylated at Tyr751 (p-PDGFRB), PDGFRB, and the loading control of GAPDH in the outer medulla. The data show individual values and mean±SEM. n = 12 per group. (d) Immunofluorescence staining of p-PDGFRB in the descending vasa recta. Red: p-PDGFRB; Blue: Hoechst 33342 (nuclei). Bar = 100 μm. ∗P < 0.05 versus the right kidney in the same group; #P < 0.05 versus the left kidney in the saline group by Steel−Dwass test. R: right kidney (control); L: left kidney (congestion). GAPDH, glyceraldehyde-3-phosphate-dehydrogenase; PDGFR, platelet-derived growth factor receptor.
Among renal function parameters, creatinine level was not different between the two groups, but blood urea nitrogen level was significantly reduced in the imatinib group compared to that in the saline group (Table S3, Supplemental Digital Content, https://links.lww.com/HJH/B967). Hepatic and cardiac dysfunction was not observed in both data of the biochemical analysis and histological analysis (Figure S4, Supplemental Digital Content, https://links.lww.com/HJH/B967).
Effect of imatinib on mRNA expressionTo clarify the mechanism by which imatinib reduced the weight of the congested kidney, we assessed the mRNA levels of genes associated with kidney injury and the PDGFR signaling pathway: Fibronectin (Fn1), α-smooth muscle actin (Acta2), Tenascin-C (Tnc), and collagens (Col1a1 and Col3a1) as fibrosis markers; Kidney injury molecule 1 (Kim1) and Osteopontin (Spp1) as kidney injury markers; Pdgfra, Pdgfrb, and Tagln as PMT markers; and Vimentin (Vim) as a marker of mesenchymal cells (Fig. 3). The mRNA expression levels of Acta2, Fn1, Kim1, Pdgfrb, Spp1, Tagln, Vim, and collagens were elevated in the cortex of the left congested kidneys. Imatinib significantly attenuated the elevation of collagens. In the outer medulla, as well as in the cortex, markers of fibrosis, kidney injury, and PMT were elevated in the congested kidneys. Imatinib significantly reduced the elevation of Tnc and Vim, and collagens. The mRNA expression of Cyclin D1 (Ccnd1), a proliferation marker, was reduced in the congested kidneys, and imatinib did not affect the reduction.
 FIGURE 3:
FIGURE 3: mRNA expression in the cortex and outer medulla. The mRNA expression level of Fn1, Acta2 (α-SMA), Tnc, Col1a1, Col3a1, Kim1, Spp1, Pdgfra, Pdgfrb, Tagln, Vim, and Ccnd1 in the cortex (a) and outer medulla (b) was assessed by qPCR. The relative mRNA expression levels were normalized to Rplp2 (ribosomal protein lateral stalk subunit P2). Data are presented as individual values and mean ± SEM; n = 10 per group. ∗P < 0.05 versus the right kidney in each group; #P < 0.05 versus the left kidney in the saline group by Steel−Dwass test. R: right kidney; L: left kidney.
Since imatinib is not a specific PDGFR inhibitor [37,40], we further assessed the mRNA expression of imatinib-binding protein kinases (Table S4, Supplemental Digital Content, https://links.lww.com/HJH/B967). Microarray data from our previous study on the creation of this renal congestion model [14] showed that the mRNA expression of some tyrosine kinases (Plk4 in the cortex, and Frk, Fyn, Mapk8, Mapk10, and Kit in the outer medulla) with lower binding affinity to imatinib than PDGFRB [37] was upregulated by the renal congestion (Table S4, Supplemental Digital Content, https://links.lww.com/HJH/B967). However, these upregulations were not replicated and imatinib did not affect the expressions by qPCR (Figure S5, Supplemental Digital Content, https://links.lww.com/HJH/B967).
Effect of imatinib on protein expressionTo further investigate the protective effect of imatinib against renal congestion, we assessed the protein expression by western blotting (Fig. 4 and Figure S6, Supplemental Digital Content, https://links.lww.com/HJH/B967). Imatinib significantly reduced the expression of ACTA2, FN1, and KIM1 in the outer medulla of the left congested kidneys (Fig. 4). Imatinib did not affect the expression of PDGFRs in the outer medulla. In contrast to the outer medulla, imatinib did not reduce the expression of those proteins in the cortex (Figure S6, Supplemental Digital Content, https://links.lww.com/HJH/B967).
 FIGURE 4:
FIGURE 4: Western blotting analysis in the outer medulla. Acta2, Fn1, Kim1, Pdgfra, and Pdgfrb were assessed by western blotting. Data are presented as individual values and mean ± SEM; n = 12 per group. ∗P < 0.05 versus the right kidney in each group; #P < 0.05 versus the left kidney in the saline group by Steel−Dwass test. R: the right kidney; L: the left kidney.
Histological changes around the vasa rectaFrom the above results, it was presumed that imatinib did not influence vasa recta dilatation and pericyte detachment, which are a trigger of kidney injury in renal congestion, but would suppress renal fibrosis and injury in the outer medulla rather than in the cortex. Therefore, we focused on the lesions around the vasa recta in histological analysis.
Histological analysis revealed hyperplasia of the extracellular matrix and renal tubular injury in the congested kidneys (Fig. 5 and Figure S7, Supplemental Digital Content, https://links.lww.com/HJH/B967). Elastica-Masson staining showed the increased fibrotic area around the vasa recta in the congested kidneys (Fig. 5a). Immunostainings of PMT markers and collagens revealed that renal congestion increased the positive area of these proteins in the interstitial space around the vasa recta. TNC, a major component of the fibrogenic niche [41], was also expressed around this area. Administration of imatinib decreased these morphological changes. Picrosirius Red staining showed an increase in collagen fibers in the congested kidney, and imatinib significantly suppressed the collagen-positive area (Fig. 5b). In contrast to the outer medulla, the extent of imatinib-induced changes in the staining intensity in the cortex was faint, similarly to the results of the qPCR and western blot. These results imply that the inhibition of the PDGFR pathway by imatinib could be beneficial against tubulointerstitial injury in our renal congestion model.
 FIGURE 5:
FIGURE 5: Histological analysis of area surrounding the vasa recta in the outer medulla. (a) Representative histological images around the vasa recta of imatinib- and saline-treated rats stained for Elastica-Masson (EM), Acta2, Tagln, and Tnc. (b) Top: Picrosirius Red staining illustrating collagen fiber around the vasa recta of imatinib- and saline-treated rats. Down: Quantification of collagen positive area. Data are presented as individual values and mean ± SEM. n = 15 (saline-treated right control kidney), 17 (saline-treated left congested kidney), 15 (imatinib-treated right control kidney), and 16 (imatinib-treated left congested kidney). Scale bar = 100 μm. ∗P < 0.05 versus right kidney in the same group; #P < 0.05 versus congested kidney in the saline group by Steel−Dwass test.
Effect of imatinib on transforming growth factor-β1 pathway and macrophage markersRecent studies revealed the cross-talk between the PDGFR pathway and TGFB pathway [22,42], and the existence of macrophage-like NG2 positive cells after renal injury [43]. The protein expression of TGFB1 and macrophage markers (CD11B, CD68, and CD206) was increased in the outer medulla after renal congestion (Fig. 6a). The phosphorylation of SMAD2/3 was also stimulated by renal congestion (Fig. 6a). Imatinib blocked the activation of the TGFB pathway and the upregulation of the macrophage markers. In double-labeling immunofluorescence staining, NG2 colocalized with PDGFRB around the vasa recta, while cells expressing macrophage markers colocalized with neither PDGFRB nor NG2 (Fig. 6b and Figure S8, Supplemental Digital Content, https://links.lww.com/HJH/B967).
 FIGURE 6:
FIGURE 6: Effect of imatinib on TGFB pathway and macrophage markers. (a) Immunoblots of TGFB1 pathway and inflammation markers in the outer medulla. TGFB1, phosphorylated (p)-SMAD2/3, SMAD2/3, CD11B, CD68, CD206, and the loading control were assessed by western blotting. The data show individual values and mean ± SEM; n = 12 per group. (b) Representative immunofluorescence images around the vasa recta of imatinib- and saline-treated rats. Scale bar = 20 μm. ∗P < 0.05 versus the right kidney in each group; #P < 0.05 versus the left kidney in the saline group by Steel−Dwass test. R: the right kidney; L: the left kidney. TGFB1, transforming growth factor-β1.
Suppressive effect of platelet-derived growth factor receptor inhibitors against pericyte-myofibroblast transition damage in vitroTo verify the effect of the PDGFR pathway inhibition against fibrogenesis via PMT, we performed in vitro experiment by using a human pericyte cell line, hPL-PC. As reported in mouse primary cultured pericytes [22], TGFB1 stimulated myofibroblast differentiation in human pericytes (Fig. 7). The expression of FN1, COL1A1, and COL4A1 was significantly elevated by TGFB1 at the transcriptional level. Both imatinib and crenolanib significantly suppressed these mRNA elevations.
 FIGURE 7:
FIGURE 7: Suppressive effect of imatinib and crenolanib against cellular fibrotic damage. The mRNA expression level of FN1, COL1A1, and COL4A1 was assessed by qPCR. The relative mRNA expression levels were normalized to ACTB and GAPDH. Data are presented as individual values and mean ± SEM; n = 8 per group. ∗P < 0.05 versus Ve; #P < 0.05 versus Ve + TGFB1 by Steel−Dwass test. Cre, crenolanib; Ima, imatinib; Ve, vehicle.
 FIGURE 8:
FIGURE 8: Schematic diagram of the present study. Renal congestion leads to vasa recta expansion, resulting in pericyte detachment. Pericyte detachment could induce platelet-derived growth factor receptors (PDGFRs) activation and pericyte-myofibroblast transition. Blocking of the PDGFR pathway by imatinib administration suppresses fibrosis around the vasa recta.
DISCUSSIONThe present study showed that the PDGFR pathway was enhanced in renal congestion and that inhibition of the PDGFR pathway by imatinib, a clinically available agent, was effective against fibrosis, even after pericyte detachment. Renal congestion is an issue of cardiorenal syndrome in heart failure. Better understandings of the pathophysiological mechanisms involved in renal congestion are needed to develop a new therapeutic strategy. Our results indicated that the activation of the PDGFR pathway after pericyte detachment was responsible for renal congestion-induced fibrosis.
In our previous study, renal congestion induced pericyte detachment which could result in extracellular matrix expansion and fibrogenesis, and the reduction of RIHP by decapsulation attenuated these injurious phenomena including PDGFRs and Tagln suppression [14]. Because renal capsulotomies are not acceptable in humans, other ways to suppress PDGFR and/or Tagln are needed. We thus assessed the effect of imatinib, inhibition of PDGFRs, against fibrosis under renal congestion. Imatinib is a potent inhibitor of multiple receptor tyrosine kinases, such as c-Abl, c-Kit, and PDGFRs, and is expected to be approved for the treatment of tumors and nonmalignant proliferative disorders [40]. Twenty mg/kg imatinib suppressed the phosphorylation/activation of PDGFRB and ameliorated renal interstitial fibrosis without hepatic, cardiac, or renal dysfunction in the present study. Imatinib at 50 mg/kg was too toxic to the kidney as concerned [44], and 5 mg/kg imatinib was not effective against congestion-derived fibrosis in our preliminary experiment.
Imatinib is not a specific inhibitor of PDGFRs, although imatinib has been used as an inhibitor of PDGFRs in animal models and is effective against kidney injury [22–26]. It has an inhibitory activity by binding to several protein kinases [37,40]. Fabian et al.[37] have identified that imatinib interacted with 16 of the 119 human protein kinases including PDGFRB, ABL, and KIT. Since the expression of PDGFRB was upregulated from the transcriptional level in the congested kidney of our renal congestion model, we further confirmed the mRNA expression of the imatinib-binding protein kinases. Their expression was not altered by renal congestion in qPCR analysis. Thus, the activation of the PDGFR pathway seems to be the major target of imatinib in the present renal congestion model. However, since we have not checked the post-transcriptional activation of these protein kinases, their contribution to renal damage after renal congestion cannot be excluded.
The consequences of abdominal venous congestion are very difficult to evaluate in humans because the concomitant reduction of renal blood flow by the low output or underlying chronic kidney diseases often occur. To dissect the direct effect of renal venous congestion, several animal experiments were performed. Cops et al.[45,46] recently reported a new rat renal congestion model with incomplete ligating thoracic IVC. In their model, kidney weight gain and renal fibrosis were not observed. This might be due to the high mortality rate of their model, as renal fibrosis can be observed only in the rats that died. In contrast, these kidney injuries were detected in our model because rats with severe renal congestion survived owing to compensation with the contralateral kidney. Furthermore, our model has the advantage that this right contralateral kidney can be the control kidney. We have shown no difference in renal function and injury between sham-operated and the right contralateral kidney in our paper on the creation of this model [14]. This would contribute to animal welfare and concise analysis of the congested kidney. It is well known that the contralateral right kidney in the unilateral ureteral obstruction (UUO) model is not normal by several factors affected by the damaged left kidney, including hemodynamic changes and neuro/hormonal transmitters [47]. This contralateral right kidney in the UUO model can only be used as a control when comparing therapeutic intervention to the obstructed kidney. Therefore, the contralateral right kidney in our renal congestion model may also need to be further validated as a control kidney.
Kidney weight gain, vasa recta expansion, and pericyte detachment in renal congestionIn the present study, the left congested kidney significantly gained weight. Inhibition of the PDGFR pathway by imatinib significantly suppressed weight gain in the congested kidney. Although cell proliferation could be activated by the PDGFR pathway [48], the expression of Ccnd1 mRNA, a cell proliferation marker and downstream of the PDGFR pathway, did not increase in the congested kidneys. Thus, this kidney weight gain was due to fibrosis and extracellular matrix expansion, but not cell proliferation. Moreover, this congestion model was different from an ischemia model with total occlusion of the renal vein, as seen in renal venous embolism, which shows severe necrotic damage (Figure S9, Supplemental Digital Content, https://links.lww.com/HJH/B967).
Vasa recta capillary expansion and pericyte detachment were observed by renal venous congestion in this study. Elevation of RIHP was observed by the increased renal perfusion pressure, but did not alter vasa recta diameter [49]. Since increased in RIHP enhanced renal oxidative stress [50], RIHP could contribute to the enhancement of the PDGFR pathway and progression of renal fibrosis by renal congestion. Indeed, TGF-β1 and NF-κB pathway were enhanced in congestive kidney, both of which promotes fibrosis with interaction with oxidative stress [14]. Furthermore, we cannot deny the possibility that renal medullary hypoxia by renal congestion may contribute to fibrogenesis in the renal medulla, which we have previously reported in Dahl salt-sensitive (Dahl S) rats, a model of cardiorenal failure [51].
In Dahl S rats, renal medullary blood flow is reduced [51]. Renal medullary fibrosis is observed following reduced medullary blood flow, increased Na reabsorption, and high blood pressure. Chiba et al.[52] has shown the close relationship between renal fibrosis and renal congestion in Dahl S rats. Therefore, the PDGFR pathway activation by renal congestion could be also responsible for renal injury in Dahl S rats. In contrast to Dahl S rats, spontaneously hypertensive rats, a genetic hypertension model, raise the blood pressure around 5–6 weeks of age, but cardiac failure and renal damages were observed at older ages [53]. This may be related to venous hypertension and renal congestion. Future studies are required to determine the roles of the PDGFR pathway in renal damage in models of hypertension.
Pathophysiological role of the platelet-derived growth factor receptor pathway in renal congestion-induced fibrosisPericytes and the PDGFR pathway normally contribute to microvessel stability and have regeneration potential [16,20,54]. Recent studies have shown that renal pericytes are major targets of kidney injurious signals, resulting in maladaptive responses and PMT with upregulation of PDGFR [20,22,43,55]. Chen et al.[22] reported that imatinib was effective against renal fibrosis in obstructive or post-ischemic kidney injury animal models with increase in platelet-derived growth factors in injured tubules and endothelium, and PDGFRα/β in interstitial pericytes and myofibroblasts. In the present study, imatinib suppressed renal medullary fibrosis without affecting the IVC pressure elevation, vasa recta expansion, and pericyte detachment. This indicates that imatinib could inhibit PMT despite maintaining renal congestion. Kittikulsuth et al.[43] demonstrated that renal NG2-positive cells had an M2 macrophage-like ability and participated in the recovery process after acute renal injury. NG2 colocalized with PDGFRB in both control and congested kidneys, but not colocalized with macrophage markers (CD11B, CD68, and CD206) in the present study. This means that the differentiation of pericytes into immune potential is limited in renal congestion, and the detached pericytes start fibrogenesis via PMT. Furthermore, a positive feedback loop between the PDGFR and TGFB pathway was reported in glomerular mesangial cells [42]. The expression of TGFB1 was elevated in the rat congested kidney, and imatinib treatment blocked the activation of the TGFB pathway in the present study. And in vitro experiment in human pericytes showed that TGFB1-induced PMT was suppressed by both imatinib and a highly selective PDGFR inhibitor. Thus, the cross-talk of the PDGFR and TGFB pathway in the pericyte could have a crucial role in renal fibrosis after renal congestion.
Efficacy of imatinib in other kidney diseasesImatinib is already commonly administered to patients with several tumors [56]. Various reports in animal experimental models of renal disorders have shown the therapeutic benefit of imatinib, including for mesangial proliferative glomerulonephritis [27], chronic allograft nephropathy [28], diabetic nephropathy [29], lupus nephritis [30,31], unilateral ureteral obstruction nephropathy [22,23,32], cryoglobulin-associated-membranoproliferative glomerulonephritis [33], and anti-glomerular basement nephritis [26]. Furthermore, Elmholdt et al.[57] reported the efficacy of imatinib against nephrogenic systemic fibrosis in humans. In these studies, the beneficial effects of imatinib are shown to be the results of its inhibitory action on the PDGFR pathway, which leads to suppression of glomerular cell proliferation or accumulation of extracellular matrix. Imatinib also reduced the number of macrophages in the nephrotoxic serum of rats with nephritis [26], suggesting the renoprotective and immunomodulatory properties of imatinib. The present study suggested that imatinib was effective against renal interstitial fibrosis induced by renal venous congestion. Administration of a specific inhibitor of the PDGFR pathway could be a promising strategy for limiting the progression of kidney diseases to end-stage renal failure.
LimitationsA few points need to be underlined as limitations of the present study. First, the present rat model artificially induces volume-overloaded venous congestion in the left kidney, as seen in the patients with congestive heart failure, by complete ligation between renal veins. Thus, this model will provide a better understanding of the pathophysiological mechanisms of renal venous congestion, but cannot be expanded to deterioration of renal function associated with progression of heart failure. Second, the IVC pressures on day 3 before tissue sampling were around 10 mmHg, which is comparable to 12.2 mmHg in Dahl S rats, a renal congestion model with heart failure [52]. However, the IVC pressure increased to around 20 mmHg at the acute phase, which may affect r
留言 (0)