Study setting
This study will take place in five ICU/HAUs in hospitals located across Canada. Participating sites include Royal Columbian Hospital, Surrey Memorial Hospital, Royal Jubilee Hospital, Nanaimo Regional General Hospital, and St. Boniface Hospital.
Eligibility criteria
The following are the inclusion criteria:
1.
ICU/HAU admission.
2.
Presence of a central venous catheter requiring locking. This includes triple lumen central venous catheters (both tunneled and non-tunneled), dialysis lines, implanted vascular access devices (IVADs), and peripherally inserted central lines (PICCs).
3.
18 years of age.
The following are the exclusion criteria:
1.
Known sensitivity to EDTA.
2.
Confirmed or suspected pregnancy, as 4% tetrasodium EDTA has not yet been investigated for use in the pregnant population.
3.
Patients who decline receiving blood products (due to the smaller blood draws and special blood conserving procedures).
4.
Physician, patient, or temporary substitute decision-maker (TSDM) declines.
5.
Currently enrolled in any other research study that may confound primary outcome measures. Co-enrollment in multiple studies will be considered on an individual basis.
6.
Patients who were previously enrolled in the study. Patients who were enrolled in the first period are not eligible for (re-)enrollment in the second period, and patients who are enrolled in the study and transferred to another participating hospital are not eligible for (re-)enrollment at the receiving hospital. Patients who had been discharged from the unit to another hospital ward and are re-admitted to the critical care unit are not eligible for re-enrollment into the study.
Who will take informed consent?
Under the guidance of the Tri-Council Policy Statement 2, this study has been approved for a waiver of consent (under Article 3.7A) by relevant ethics boards for the following reasons: 4% tetrasodium EDTA is considered at least as safe as the standard of care and has Health Canada approval, there is a strong chance that 4% tetrasodium EDTA will be safer than the standard of care, there have been no adverse outcomes reported in previous clinical use, and the study could not be feasibly completed with a deferred or a full consenting process due to the rapid decision to use a CVAD lock and the need to use it immediately. Due to practical barriers caused by the volume of patients enrolled in the study, it will not be possible for the research staff to inform all patients of their participation in the clinical trial once treatment is finished.
Reminders and signage relevant to the trial will be present in each unit during study procedures. Should patients or caregivers have questions about the trial, a patient/caretaker information letter will be provided along with contact information for the research coordinator. Patients/caretakers will be informed that they can remove themselves or their family members from the trial at any time, with no consequence to their care.
Additional consent provisions for collection and use of participant data and biological specimens
This trial does not involve collecting biological specimens for storage (see item ).
InterventionsExplanation for the choice of comparators
The comparator to the trial intervention will be the standard of care CVAD lock. This includes saline for central venous catheters and peripherally inserted central catheters, and citrate for hemodialysis catheters. As the aim of the study is to determine whether 4% EDTA is more efficacious in preventing CVAD complications than the current standard of care, we must compare this alternative to current practices.
Intervention description
Upon admission to the ICU/HAU, any patient with a CVAD requiring locking who meets the criteria will be enrolled in the study. The patient will receive either a standard of care lock or a 4% tetrasodium EDTA locking solution depending on the randomization status of their treating hospital. Syringes containing either 4% tetrasodium EDTA solution or standard of care locking fluid to be used during locking procedures (called “locking kits”) will be prepared ahead of time by the research pharmacy according to the randomization status of the unit. They will be clearly labeled for use as a locking solution only and placed at the patient’s bedside. Syringes will be prepared with an additional label to remind nurses not to use them in pregnant patients or in those who decline any blood products. In addition to the bedside assessment, research coordinators will review each patient that has received a locking solution every day to gather data and to ensure compliance with the study protocols. Nurses will use the prepared syringes to lock and maintain each CVAD according to the standard of care. Syringe supply at each patient’s bedside will be replenished as needed by the research pharmacy and research coordinators. Enrollment can occur 24 h per day, 7 days per week as nurses can obtain locking syringes specific to the randomization status of the unit at all times.
Medication, maintenance fluids, pressors, or anesthesia will be administered through the CVAD according to the standard of care. Once a lumen of the catheter is no longer being utilized for continuous infusions, leftover medication will be aspirated and flushed with 20 mL of 0.9% sodium chloride using a turbulent flush method as per the standard of care. For patients randomized into the experimental group, the catheter will then be locked with the appropriate volume of 4% tetrasodium EDTA, as specified by catheter manufacturer instructions. These procedures will remain exactly the same for patients randomized into the control group with the exception of using saline or citrate as a locking fluid. Experimental and control solutions are indistinguishable by visual inspection.
All further catheter procedures and maintenance will proceed according to the standard of care. For all locked catheters, the frequency of flushing will occur every 12 h (or 72 h for dialysis catheters). During this procedure, catheters locked with the standard of care lock will be aspirated, flushed with 10 mL of 0.9% sodium chloride using turbulent flow, and re-filled with the appropriate volume of the standard of care lock. Procedures will stay exactly the same for patients in hospitals randomized to administer the 4% tetrasodium EDTA treatment, except for continued locking with 4% tetrasodium EDTA. Neutral displacement cap change procedures will remain unchanged in both groups. This includes a change every 96 h, after a blood draw through a cap, if removed, contaminated, damaged, and as needed.
Regular assessments of the site of insertion, CVAD system, and patency will continue according to the standard of care, regardless of the type of locking solution used. According to the adult CVAD maintenance record used at each hospital, nurses will assess that dressing is secure, dry, and intact and will palpate the site and check the system at the beginning of each shift. The site of insertion and connections will be reassessed every 4 h as is standard protocol. Dressing and tubing changes will also proceed according to the standard of care in both groups.
Should a locked line (locked with either standard of care or 4% tetrasodium EDTA solution) needs to be re-opened for additional medication administration, the line will be assessed for patency, aspirated of locking solution, and flushed with 20 mL of 0.9% sodium chloride before administering further medication. Patency will be assessed by the ability to aspirate for blood return and the ability to flush a CVAD without resistance prior to the administration of medications and solutions. If the line is not patent, the nurse will assess for occlusions and proceed with the standard of care occlusion diagnosis and treatment. As per the standard of care, CVADs will continue to be removed at the earliest opportunity for each patient. Four percent of EDTA has been shown to be safe if it is inadvertently flushed into the patient, thereby ensuring an adequate safety profile for use with these vulnerable patients.
Criteria for discontinuing or modifying allocated interventions
The patient may withdraw from the trial at any time. Patients for whom an exclusion criterion is discovered after enrollment will be withdrawn from the study and excluded from the analysis.
Strategies to improve adherence to interventions
The patients themselves do not have to do anything to adhere to the intervention or provide measurements of primary and secondary outcomes. The intervention itself will be administered by the ICU/HAU staff at each center. Strategies to ensure the staff adhere to the experimental procedures include education sessions on the purposes and procedures of the trial before the start of the experiment, and during the trial. The ICU/HAU staff have also been a part of the protocol development to ensure a seamless incorporation of experimental procedures into the standard workflow. The primary and secondary outcomes are already captured as part of the standard of care and will be recorded in patient charts. These charts will be reviewed by the research staff to fill out the study case report forms (CRFs).
Relevant concomitant care permitted or prohibited during the trial
Clinical care will continue as per the discretion of the patient’s healthcare team. The intervention is a CVAD locking fluid that remains only in the lumen of the CVAD; it will never actually enter the patient’s circulation. The locking fluid will be aspirated out of the catheter following the end of locking. As such, there is no potential for drug interactions, and we do not foresee any restrictions in enrolling patients based on other types of concomitant care.
Provisions for post-trial care
There is no anticipated harm and compensation for trial participation.
Outcomes
The following is the primary outcome:
1.
Incidence rate of the following: confirmed CLABSI as diagnosed in accordance with the Fraser Health CLABSI diagnosis algorithm (see Additional file 1) based on the NHSN guidelines, CVAD replacement due to occlusion, and/or catheter obstruction requiring alteplase use. The composite incidence rate will be calculated as the number of events divided by the number of catheter days. The patient is considered at risk during the time the patient is in the ICU/HAU and has a catheter placed. Multiple outcomes occurring within a 24-h time frame will be counted as a single event, except for CLABSI; any occurrence less than 2 calendar days following a previous CLABSI and subsequent catheter removal will be considered attributed to the previous catheter, and treated as a single event. The occurrence outcomes will be monitored twice daily via reviewing patient charts and participating with discussions with the patients’ treating nursing staff. Outcomes will be tested for and diagnosed according to clinicians in charge of patient care.
Note about the primary outcome
In the case of suspected CLABSI, diagnostic procedures will proceed according to the Fraser Health CLABSI Case Identification Algorithm, in all participating ICU/HAUs. Following CLABSI diagnosis, patients will receive antibiotic treatment as necessary. Should the care team decide that replacement of the CVAD be necessary, treatment will proceed according to the standard of care. Upon subsequent locking procedures, patients will receive the same locking solution as the rest of the hospital unit at that particular point of the study procedure.
The decision to measure CLABSI as an outcome instead of CRBSI was made to keep with the real-world, pragmatic design of the trial. Diagnosis of CRBSI would require specific research-related procurement of samples and microbiological testing that due to the sheer size of the trial, would be too expensive and difficult to implement logistically. Additionally, the use of the CLABSI outcome enables a comparison of rates of catheter-related infections of ICUs participating in this study to those participating in the Canadian Nosocomial Infection Surveillance Program [30]. This surveillance program collects hospital-acquired infection data from 40 participating hospitals across Canada and serves to inform governments and policymakers of the burden of hospital-acquired infections. We chose to also report the same outcome so that the results of our study could be more widely compared to the levels of CLABSI across other Canadian ICUs.
To keep with the current standard of care, CVADs will only be tested for infection at the discretion of the treating physician and will not be tested systematically. This is another factor that we feel justifies the use of CLABSI over CRBSI, although we recognize the outcome is not as rigorous as the CRBSI diagnosis and may be considered a limitation of our work.
Secondary outcomes
All outcomes will be assessed twice daily via reviewing the patient charts and participating in discussions with the patients’ treating nursing staff. The outcomes will be tested for and diagnosed according to clinicians in charge of patient care.
1.
Incidence rate of confirmed CLABSI (as adjudicated according to the NHSN criteria described previously).
2.
Incidence rate of suspected CLABSI. A case of suspected CLABSI will be defined as a case in which no microbiological testing was performed on the suspected catheter or through-catheter blood sample, with the patient having a positive blood culture result of a common bacteria associated with a CVAD infection with no other identifiable source of infection.
3.
Incidence rate of catheters requiring removal due to occlusion, as determined by inspecting relevant notes on the patient chart and discussion with the nursing staff.
4.
Incidence rate of catheters requiring alteplase use as determined by inspecting relevant notes on the patient chart and discussion with the nursing staff.
5.
Incidence rate of catheter-associated venous thrombosis, as determined by inspecting relevant medical imaging and notes on patient charts.
6.
Incidence rate of catheter colonization, as diagnosed according to the Clinical Practice guidelines of Infectious Diseases Society of America [31].
7.
Classification of types of microorganisms isolated from a convenience sample of removed CVAD’s through culture-dependent and independent techniques.
8.
Health economic evaluation of the cost of intervention versus health care utilization and associated costs of diagnosis and treatment of catheter complications
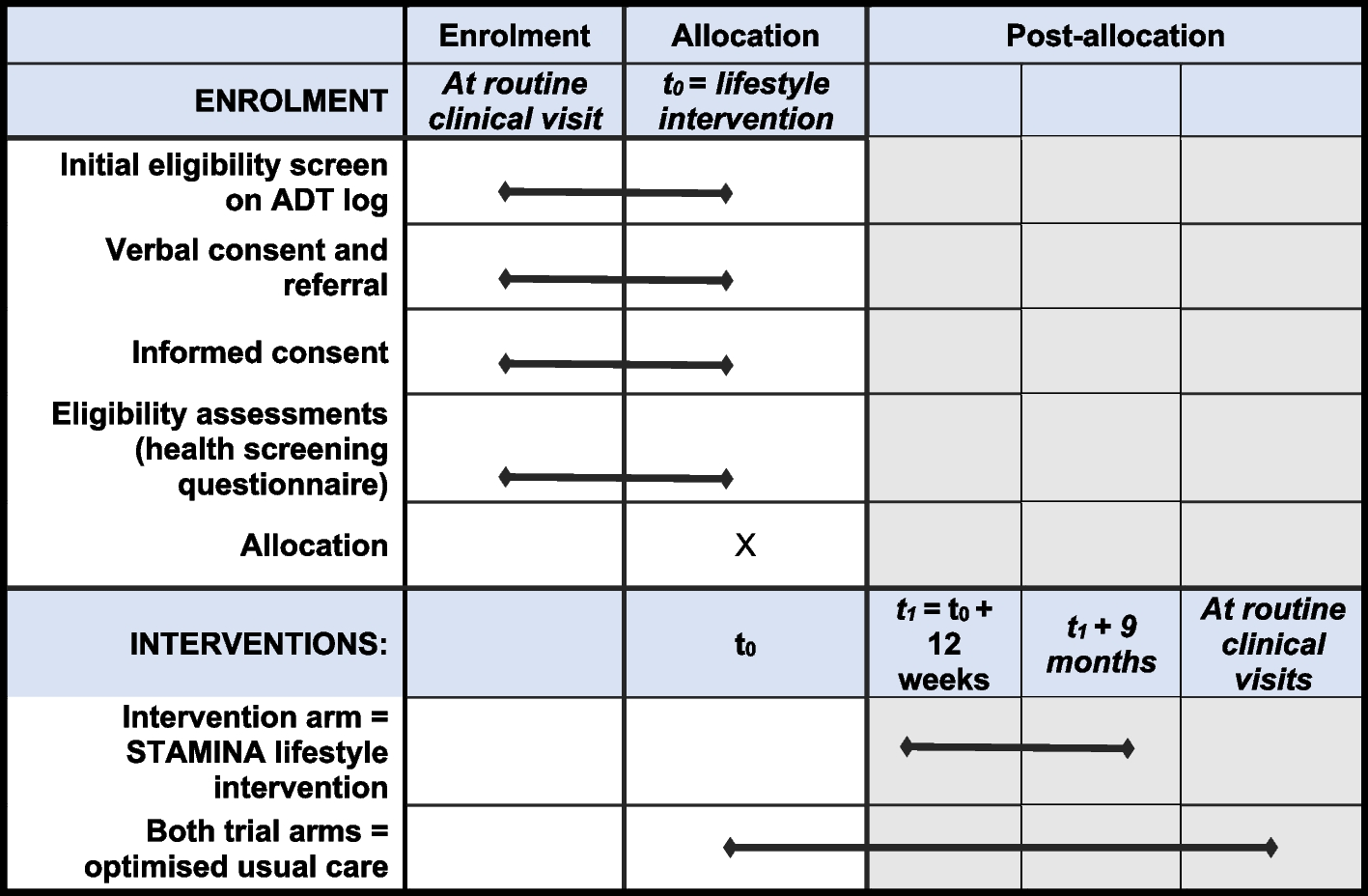
Participant timeline
Participant timeline is described in Table 1.
Table 1 Participant timelineParticipants will be enrolled during the first three-and-a-half months of each four-and-a-half--month period. Each participant will be followed until they leave the ICU/HAU, they reach the end of the 28-day follow-up period, or they die. Data collection for incidence rates of confirmed/suspected CLABSI, catheter occlusion requiring removal, catheter obstruction requiring alteplase use, direct cost related to alteplase use for catheter occlusion, and catheter colonization will continue via the combination of in-person discussion and chart review for the duration of time that the patient has their catheter in place, until they are discharged from the ICU/HAU, or they die, whichever comes first. In the event that a blood culture is taken on either the last day of catheter placement or the last day that the patient is in the unit, the research coordinator will continue to monitor the patient charts for the result of that blood culture, as a bloodstream infection may still be attributed to the catheter.
Data collection in the form of monitoring medical imaging results for the incidence rate of catheter-associated venous thrombosis will continue for the entirety of the patients’ ICU/HAU stay, even if all CVADs have been removed. The senior investigators’ clinical experience indicates that thrombosis from a CVAD in the upper thoracic region is sometimes not discovered until weeks after the CVAD has been removed, as it may take this long for the patient to develop symptoms of thrombosis. As such, the research coordinators will continue to follow the medical imaging results of the patients who are still in the ICU/HAU but have had all central lines removed and are no longer receiving the study intervention.
Sample size
The results from a 1-month observational study conducted at Royal Columbian Hospital indicate a 20% baseline risk of the composite outcome [32]. The analysis included central venous catheters, PICC lines, and hemodialysis catheters. In previous clinical studies, 4% EDTA solution has shown a 40–70% reduction in occlusion (measured by total alteplase use before vs. after intervention). Assuming a conservative 30% relative risk reduction in the risk of the composite outcome, our sample size calculation is based on an absolute risk reduction from 20 to 14%. A total of 1690 patients need to be recruited to achieve adequate power.
The simulated sample size calculations were performed using R with 10,000 simulation runs, which yields a standard error of 0.4% in the estimated power. The calculation assumed a standard deviation of baseline risk across different ICUs to be 0.02, which corresponded to a within-cluster within-period correlation of 0.0029. The standard deviation between cluster time periods was assumed to be 0.001. A generalized linear mixed effect model was used to generate and analyze the data. The statistical significance was determined using a t-test with degrees of freedom equal to the number of cluster periods minus the number of periods minus one [33].
Recruitment
The results of the observational study conducted at Royal Columbian Hospital’s 30-bed ICU and step-down unit in December 2020 reveal that 75 patients out of a total of 90 met the inclusion criteria to be enrolled in the study. Conservatively, we aim to recruit at least 50 patients per unit per month at each participating unit. We do not perceive recruitment challenges as we have obtained waived consent from relevant ethics boards.
With a three-and-a-half-month enrollment window during each period, each site is expected to recruit at least 175 patients per period. As five ICUs will be participating in this study and each site enrolls for two periods, the enrollment will be 1750 patients in total. Protocol adherence will be deemed a success if there is greater than 85% of patients who are adherent to the entire study protocol.
Assignment of interventions: allocationSequence generation
Randomization involves only the allocation of the five ICUs to the locking solution that will be used during the first period. The randomization will be performed by a statistician at the Centre for Health Evaluation and Outcome Services (CHEOS).
Concealment mechanism
The statistician performing the randomization will disclose the allocations to only the study project manager (PM). The PM will then liaise with each site pharmacy to inform them of their experimental condition so that they can prepare the appropriate locking kits during each period. Blinding of clinicians and research staff will remain throughout the entirety of the trial.
Implementation
Allocation sequence will be generated by the external statistician. Study participants will be enrolled by designated research coordinators at each site. All patients at each participating site will receive the treatment which is being administered at the site during that period.
Assignment of interventions: blindingWho will be blinded
Trial participants, care providers, outcome assessors, and data analysts will be blinded to the assignment of intervention. The only research personnel who will be unblinded are the statistician performing the randomization, the PM, and the research pharmacists in charge of supplying locking kits used during the study. As each of the 4% tetrasodium EDTA, saline, and citrate will be placed in 10mL syringes, they will not be distinguishable to the trial participants, care providers, or outcome assessors to maintain their blinding. Data analysis will occur outside of the unit with the type of intervention received by each patient concealed until the completion of analysis.
Procedure for unblinding if needed
If unblinding is necessary for patient management (e.g., in the case of an adverse event (AE) for which patient management requires knowledge of treatment assignment), the investigator will be able to obtain this information by getting approval from the study principal investigator (PI) to contact a designated individual with access to the allocation list. The study PI and the designated individual will be available 24 h per day and 7 days per week. Treatment codes will not be broken except in emergency situations that affect clinical care decisions. The investigator requesting unblinding will document and provide an explanation for the request.
Data collection and managementPlans for assessment and collection of outcomes
All information collected for each patient will be obtained either by reviewing patient charts or engaging in conversations with the treating nurses daily. Members of the research team at each site will extract each participant’s study data from the patient charts and/or resulting nursing conversations and enter the data directly into a secure, electronic database. The lead investigator will instruct each of the study sites regarding data capture procedures on electronic and/or paper CRFs.
Plans to promote participant retention and complete follow-up
All experimental procedures will be administered by ICU/HAU and the research staff during the patient stay. As such, there will be no efforts to promote participant retention.
Patients will be followed by the members of the research team until they leave the ICU/HAU, they reach the end of the follow-up period, or they die. After leaving the ICU/HAU, the patients will no longer be part of the study and will not be retained. Patients who are transferred out of the ICU/HAU with a central line in situ will no longer receive the study locking solution and revert to the standard of care for the unit to which they have been transferred to. To avoid logistical complications due to the sheer number of participants, patients who leave the ICU/HAU for another hospital unit with their original CVAD still inserted will not be followed. Patients will not be followed past the end of the experimental period.
Data management
Data will be managed by an external data monitoring center. The secure database will be managed according to the standard operating procedures and will remain in secure locations on site throughout the entirety of the study.
Confidentiality
In order to maintain patient privacy, data capture tools, study drug accountability records, study reports, and communications will identify the assigned patient number. Confidentiality standards are maintained by coding each patient enrolled in the study through the assignment of a unique patient identification number. Patient names or any identifying information will not be included in the aggregate data that are transmitted for analysis. Only site enrollment logs maintained by research coordinators in charge of data acquisition will contain any personal identifiers. These logs will remain in a secure location on site for the duration of the study. Only research coordinators will access this information. Additionally, the site investigators will grant monitor(s) and or its designee access to the patient’s original medical records, including medical history, laboratory studies, and medication administrations, for verification of data gathered and auditing the data collection process. This information will be accessed for the duration of the study for the purpose of data verification and reconciliation.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use
A sample of convenience (n = 15) of all catheters removed for suspected or confirmed CLABSI or colonization during work hours at the lead investigators site will be collected and used for the analysis of biofilm formation and classification of microorganisms. The same number of CVADs removed from patients who no longer require a CVAD with the absence of colonization will also be collected to act as a control comparator in this analysis. Upon removal, these CVCs will be placed in specimen bags stored at 4 °C. These catheters will then be processed by study personnel before shipment to Dr. Aaron White’s lab at the Vaccine and Infectious Disease Organization-International Vaccine Centre (VIDO-InterVac) for further analysis. These will include culture-dependent and culture-independent classification of microorganism colonies and analysis of biofilm, according to internal standard operating procedures.


留言 (0)