
記住我
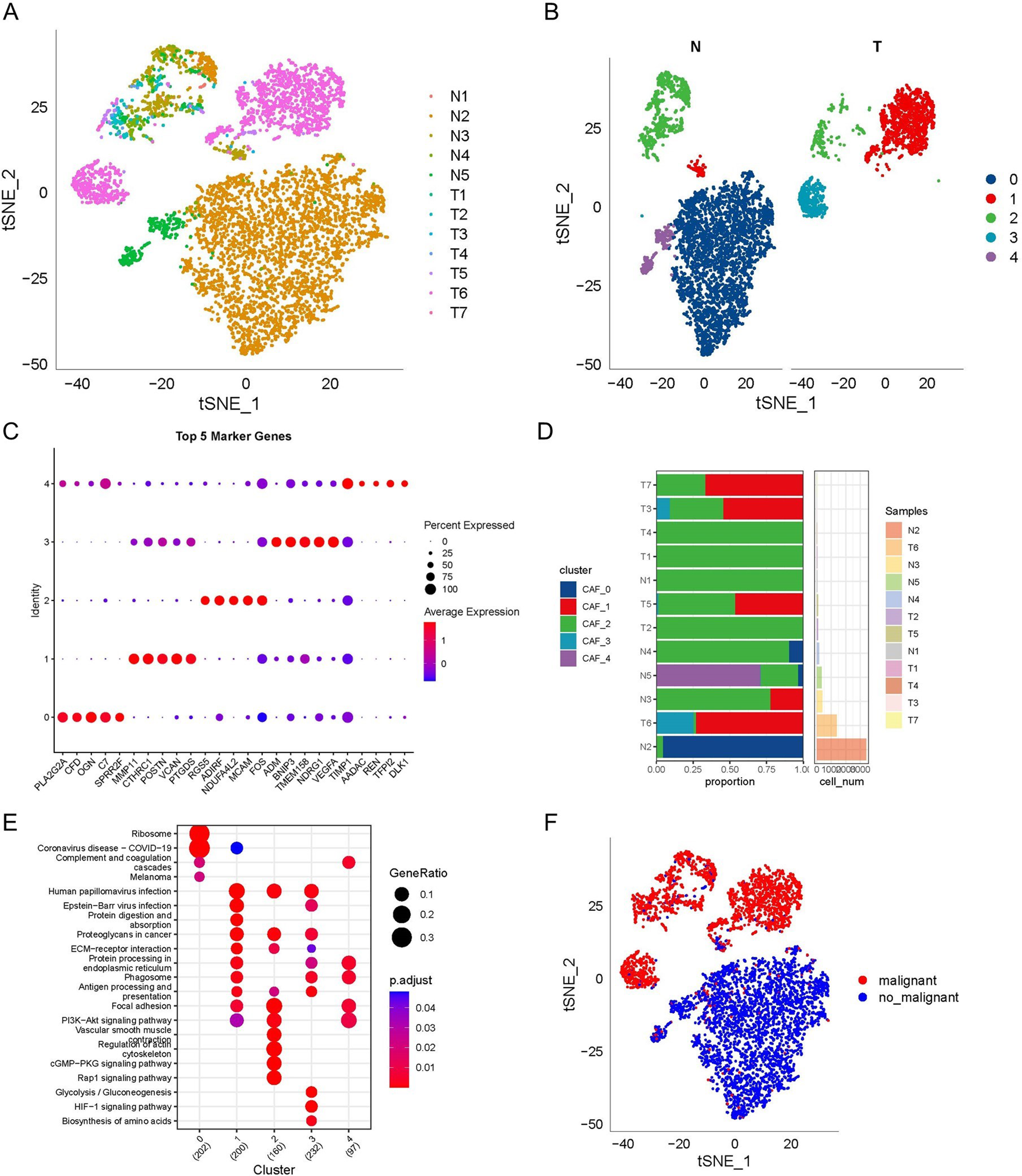
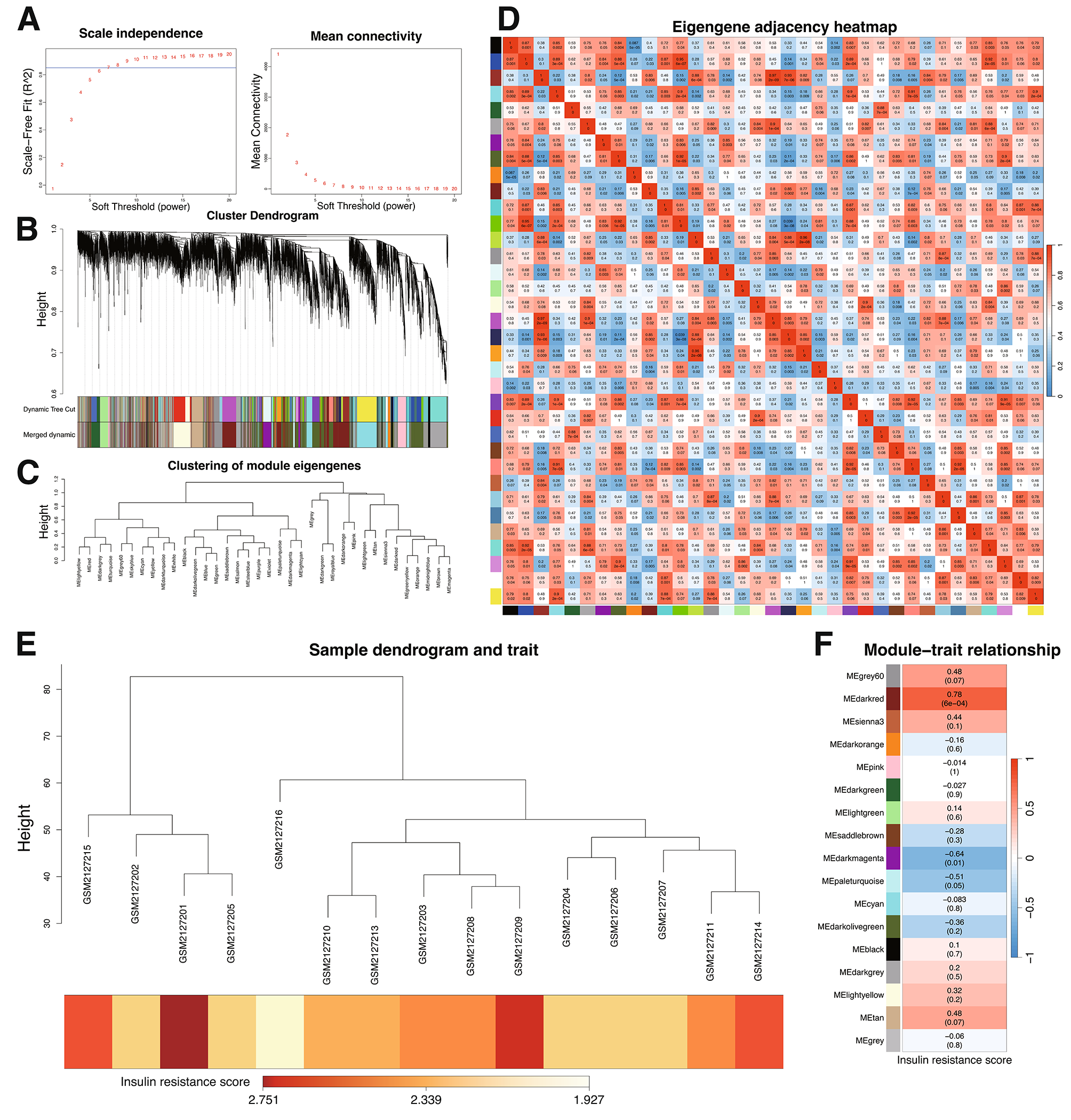

In this study, sphere-forming cells derived from A2780 and SKOV3 epithelial ovarian cancer cells, termed A2780-SP and SKOV3-SP cells, respectively, were used as ovarian CSCs to evaluate the role of circRNAs in ovarian CSCs. As shown in Fig. 1A, the spheres were cultured for 7 days, and then images showing morphology of spheres were taken under the phase contrast microscope. To identify the expression of circRNAs in ovarian CSCs, microarrays based on circRNAs were conducted on ovarian CSCs (A2789-SP, SKOV3-SP) compared to monolayer cells (A2780 and SKOV3 cells). The box plot represents that the median intensity values in ovarian CSCs (A2780-SP, SKOV3-SP) and monolayer cells (A2780, and SKOV3) were almost similar after normalization (Fig. 1B). As shown in Fig. 1C, hierarchical clustering demonstrates the expression of multiple circRNAs in the CSCs (A2789-SP and SKOV3-SP) and monolayer cells (A2780 and SKOV3 cells). The scatter plot and volcano in Fig. 2A and B shows the differences in circRNA expression between CSCs and monolayer cells. Our results showed that 159 circRNAs were significantly upregulated and 55 circRNAs were significantly downregulated in ovarian CSCs compared with the monolayer cells (fold change > 1.5) (Supplementary material). In A2780-SP cells, 2447 circRNAs were upregulated and 2346 circRNAs were downregulated compared to A2780 (log2 (fold change)| > 1.5 and a P-value < 0.05 were used to evaluate significant differences in the expression of circRNAs between the two groups) (Supplementary material). In SKOV3-SP cells, 2447 circRNAs were upregulated and 22,982 circRNAs were downregulated compared to SKOV3 cells (Supplementary material). The top 10 upregulated or downregulated circRNAs in ovarian CSCs compared to monolayers are listed in Table 1. In addition, Circ_004766, Circ_008603, Circ_004908 were downregulated in ovarian CSCs compared to control (supplementary material). Consistent with our data, these circRNAs were also downregulated in the ovarian cancer tumor tissues compared to normal ovary tissues using public dataset (GSE192410) (Fig. 2E).
Fig. 1
Differentially expression of circRNAs in ovarian cancer stem cells. A A representative image of ovarian cancer stem cell sphere formation originating from A2780 (upper) and SKOV3 (lower) cells on day 7 of culture. B The box plot shows variations in circRNA expression in ovarian cancer stem cells derived from A2780 and SKOV3 cells compared to control. C Heat map of the circRNA microarray profiles representing the expression of circRNAs between the adherent cells (A2780, SKOV3) and cancer stem cells (A 2780-SP, SKOV3-SP). Green color showing lower expression levels and red color indicating higher expression levels
Fig. 2
Distributions of circRNAs in human chromosomes in ovarian cancer stem cells. A Scatter plots representing differentially expressed circRNAs in ovarian cancer stem cells. circRNAs above and below the border green line show more than twofold change in expression. B The volcano plots representing differentially expressed circRNAs between groups. The green vertical line marks twofold changes, while the horizontal line represents a P-value of 0.05. Distribution of differentially upregulated (C) and downregulated D circRNAs between ovarian cancer stem cells and monolayer cells in human chromosomes. The ratio of circRNAs originated from exonic, intronic, and intergenic regions are shown. E The circRNA_004766, circRNA_008603, circRNA_004908 level of the GSE192410 dataset, which contains information on ovarian cancer tumour tissue
Table 1 The top 10 upregulated and downregulated circRNAs from ovarian cancer stem cells compared to monolayer cellsExpression of circRNAs in human chromosomesAmong all differentially expressed circRNAs, 159 upregulated and 55 downregulated circRNAs overlapped between A2780-SP and SKOV3-SP cells. Of the 159 upregulated circRNAs, 122 (76.7%) were transcribed from the exonic region, 14 (8.8%) from the sense-overlapping region, 21 (13.2%) from the intronic region, and 2 (1.2%) from the intergenic region (Fig. 2C), whereas of the 55 downregulated circRNAs, 53 (96.3%) were transcribed from the exonic region, 1 from sense-overlapping region (1.8%), and 1 from the antisense region (1.8%) (Fig. 2D).
GO functional annotation and pathway enrichment analysis of differentially expressed circRNAscircRNAs are encoded by their parental genes, and one of their primary functions is to regulate parental gene expression [17]. To identify the function of circRNAs in ovarian CSCs, we performed GO functional pathway enrichment analysis of parent genes of differentially expressed circRNAs. The 10 most significant GO terms of the upregulated circRNAs in ovarian CSCs are listed in Fig. 3. The identified BP terms were localization, regulation of response to stimulus, regulation of signaling, anatomical structure morphogenesis, cell morphogenesis, regulation of catabolic process, histone modification, covalent chromatin modification, and histone H3-K9 modification (Fig. 3A). The identified MF terms were nucleoside phosphatase binding, carbohydrate derivative binding, nucleotide binding, purine ribonucleoside triphosphate binding, purine ribonucleotide binding, purine nucleotide binding, adenyl ribonucleotide binding, adenyl nucleotide binding, ATP binding, and PDZ domain binding (Fig. 3B). The identified CC terms were intracellular, organelle, membrane-bounded organelle, intracellular organelle, cytoplasm, intracellular membrane-bounded organelle, cytosol, and actin cytoskeleton (Fig. 3C).
Fig. 3
GO analysis for upregulated circRNAs in ovarian cancer stem cells (A2780-SP, SKOV3-SP) compared to monolayer cells. A for BP; (B) for CC (C) for MF. BP: biological process; CC: cellular component; MF: molecular function
The 10 most significant GOs of the downregulated circRNAs are listed in Fig. 4. The identified BP terms were cellular macromolecule metabolic process, cellular protein metabolic process, cell cycle, apoptotic signaling pathway, extrinsic apoptotic signaling pathway, and activation (Fig. 4A). The MF terms were protein binding, transferase activity, transcription corepressor activity, modification-dependent binding, translation regulator activity, nucleic acid binding, translational regulator activity, peptide N-acetyltransferase activity, tumor necrosis factor receptor superfamily binding, translation factor activity, RNA binding, N-acetyltransferase activity, and translation initiation factor activity (Fig. 4B). The identified CC terms were intracellular, organelle, membrane-bounded organelle, nucleus, nucleoplasm, condensed chromosome, acetyltransferase complex, protein acetyltransferase complex, N-terminal protein acetyltransferase complex, and nuclear pore outer ring (Fig. 4C).
Fig. 4
GO analysis for downregulated circRNAs in ovarian cancer stem cells (A2780-SP, SKOV3-SP) compared to monolayer cells (A2780, SKOV3). A for BP; (B) for CC; (C) for MF. BP: biological process; CC: cellular component; MF: molecular function
Next, to investigate the functional roles of circRNAs in CSCs, the KEGG pathway was used with mRNAs transcribed from the parent genes of differentially expressed circRNAs in ovarian CSCs. From the upregulated and downregulated circRNAs in ovarian CSCs, the 10 most significant KEGG pathways are listed in Fig. 5 A and B. As shown in Fig. 5A, KEGG analysis of upregulated circRNAs indicated enrichment in glutamatergic synapse; long-term potentiation; alanine, aspartate, and glutamate metabolism; GABAergic synapse; salivary secretion; valine, leucine, and isoleucine degradation; MAPK signaling pathway; Fc gamma R-mediated phagocytosis; Notch signaling pathway; and endocrine and other factor-regulated calcium reabsorption. KEGG analysis of downregulated circRNAs revealed enrichment in cell cycle, ubiquitin-mediated proteolysis, RNA transport, DNA replication, lysine degradation, protein processing in endoplasmic reticulum, mRNA surveillance pathway, fanconi anemia pathway, homologous recombination, and mismatch repair (Fig. 5B).
Fig. 5
KEGG pathway analysis for upregulated or downregulated circRNAs in ovarian cancer stem cells (A2780-SP, SKOV3-SP) compared to monolayer cells (A2780, SKOV3). KEGG pathway analysis shows the top 10 enriched pathways for upregulated A and downregulated (B) circRNAs in the ovarian cancer stem cells
Survival analysis of parental gene expression in ovarian cancer cellscircRNAs are encoded by their parental genes, and one of the primary functions of circRNAs is to regulate parental gene expression [17]. To understand whether these parental genes are related to ovarian cancer, the association between the expression of the parental genes of the top 10 circRNAs and survival was examined. We found that the expression of six genes (BCLAF1, FBLN1, ARHGAP23, STON2, UBQLN4, and ATP2B1) had a significant positive correlation with the survival rate of patients with ovarian cancer (Fig. 6).
Fig. 6
Survival analysis of expression of parental genes (BCLAF1, FBLN1, ARHGAP23, AHNAK2, STON2, AACS, INCENP, UBQLN4, RRM2B, ATP2B1) in ovarian cancer cells
Prediction of common MREs of differentially expressed circRNAs and mRNAsIt has been reported that circRNAs can sequester relevant miRNAs through MREs to post-transcriptionally regulate gene expression [18]. Therefore, we investigated potential miRNA targets of the validated circRNAs using miRNA target prediction software. The putative MREs of differentially expressed top 10 upregulated and downregulated circRNAs are listed in Tables 2 and 3.
Table 2 Prediction of miRNA binding sites on the top 10 upregulated circular RNAsTable 3 Prediction of miRNA binding sites on the top 10 downregulated circular RNAsBioinformatics analysis of validated circRNA-miRNA-mRNA networksTo examine circRNA-miRNA-mRNA networks, we chose hsa-circRNA000963, which is highly expressed in ovarian CSCs. hsa-circRNA 000963 has five potential miRNA targets (hsa-miR-629-3p, hsa-miR-298, hsa-miR-424-5p, hsa-miR-497-5p, and hsa-miR15b-5p). The miRNA walk database was used to predict miRNA targets. As shown in Fig. 7A, a Venn diagram was drawn to identify overlapping targets among five miRNAs (hsa-miR-629-3p, hsa-miR-298, hsa-miR424-5p, hsa-miR-497-5p, and hsa-miR-15b-5p). To determine the function of the target, we selected the target from at least two overlapped miRNAs among the five miRNAs that were used for bioinformatics analyses. KEGG pathway analysis for 425 target miRNAs showed that they were involved in hsa04550: signaling pathways regulating pluripotency of stem cells, hsa04151: PI3K-Akt signaling pathway, hsa04110: cell cycle, hsa04114: oocyte meiosis, hsa04115: p53 signaling pathway, and so on (Fig. 7B). GO enrichment analysis showed that GO terms were revealed in GO:0007223 ~ Wnt signaling pathway, calcium modulating pathway, GO:0035196 ~ production of miRNAs involved in gene silencing by miRNA, GO:0035280 ~ miRNA loading onto RISC involved in gene silencing by miRNA, GO:0000122 ~ negative regulation of transcription from RNA polymerase II promoter, GO:0000082 ~ G1/S transition of mitotic cell cycle (Fig. 7C).
Fig. 7
GO and KEGG pathway analysis for the mRNAs in the circRNA-miRNA-mRNA network. A A Venn diagram was drawn to identify overlapping targets among five miRNAs (hsa-miR-629-3p, hsa-miR-298, hsa-miR424-5p, hsa-miR-497-5p, and hsa-miR-15b-5p) which possess MRE binding site for hsa_circRNA_000963. KEGG pathways (B) and GO terms (C) for the target genes are shown. Targets from at least 2 overlapped miRNAs among 5 miRNAs were used for bioinformatics analyses
留言 (0)