This SAP describes the statistical analyses planned for the NOTACS trial. The health economic analyses are described separately in a health economics plan.
Statistical principles
This SAP is based on version 3.0 (19 January 2022) of the trial protocol and version 0.43 (13th June 2022) of the trial SAP. The statistical analyses will be carried out using R (www.r-project.org). Other major statistical software may be used where appropriate. Derived variables are described in the additional document. Data will be checked for outliers and unexpected distributions or data points will be queried. Consistency checks between two or more variables will also be performed. Where relevant, variables will be summarised by the treatment arm using the following descriptive statistics: for continuous variables, the non-missing sample size, mean, standard deviation, median, maximum and minimum; for categorical variables, the frequency and percentages (based on the non-missing sample size) of observed levels will be reported.
Handling of missing data
Withdrawal rates will be summarised by the treatment arm, and the timing of and reason for withdrawals will be reported. The proportion of missing data will be quantified by the treatment group for the variables included in the primary and secondary analyses. Based on the pilot study, the rate of missing data in DAH90 is expected to be low. Variables with >25% missing data may not be used in statistical regression modelling.
The trial was powered on the basis of a 5% loss to follow-up rate. Provided there is no more than 5% missing data in DAH90 and no obvious cause for concern over the pattern of missing data then we will run complete-case analysis. If the missingness rate in DAH90 exceeds 5%, we will take further steps to investigate the type of missingness for the subset of variables included in the primary and secondary analyses. If the missing data is found not to be MCAR in any instance, we will assume the missing data is MAR and perform multiple imputation as a sensitivity analysis to evaluate the robustness of the primary analysis. Our approach to assessing missing data and implementing multiple imputation is detailed in the additional document.
For derived variables such as DAH, the presence of missing data in the variables used to calculate these from may be more difficult to identify. DAH relies on participant location diaries to capture all changes in participant location with accurate start and end dates for each change in location. There may be instances where data is missing from participant location diaries despite it appearing that the participant location diary has been completed in full, if for instances a participant fails to record a change of location. This is a potential limitation of the DAH end-point. Where it is clear that a patient diary is incomplete, they will be completed where possible by calling the patient’s GP surgery and using hospital discharge summaries.
Patient flow
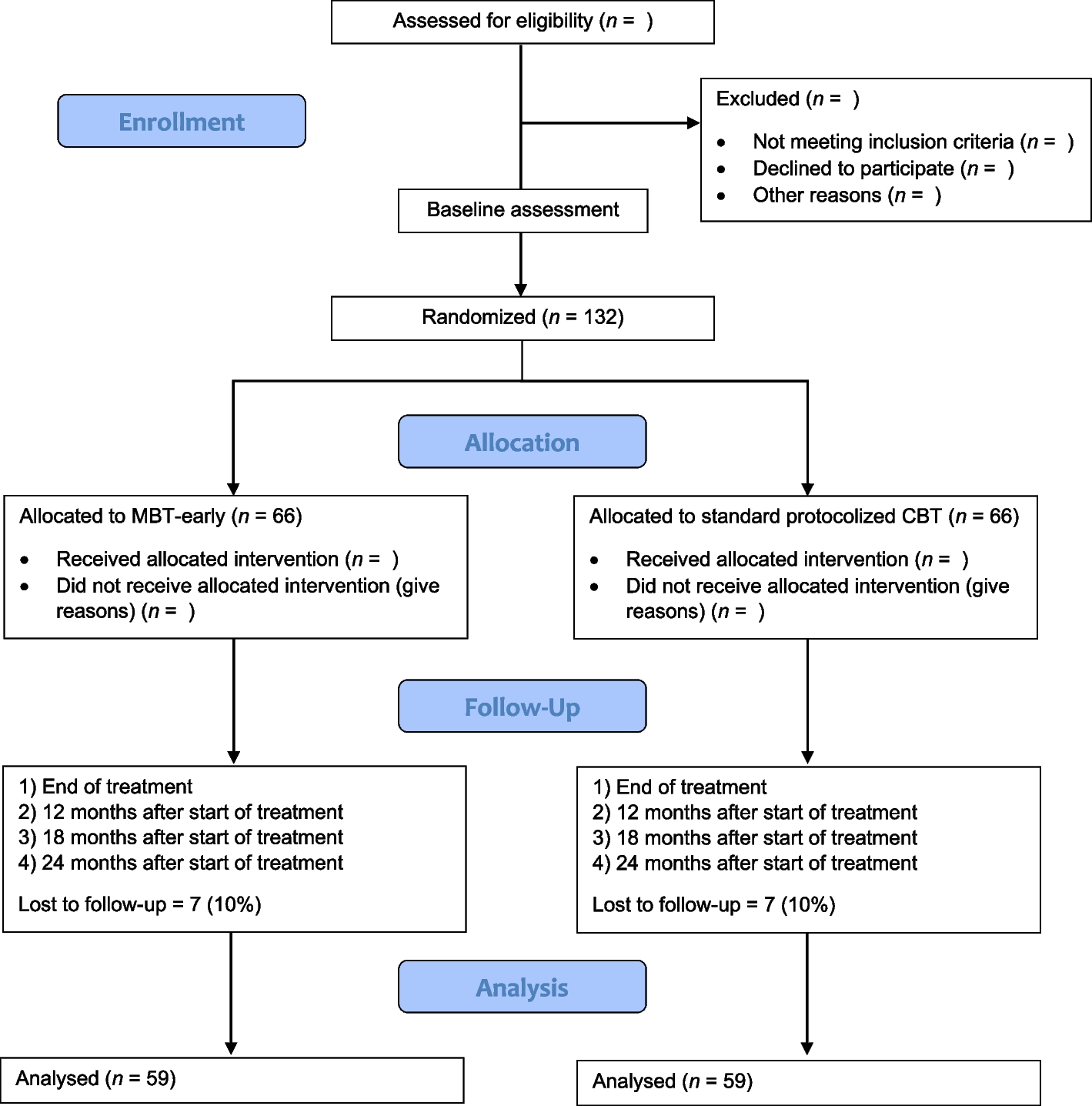
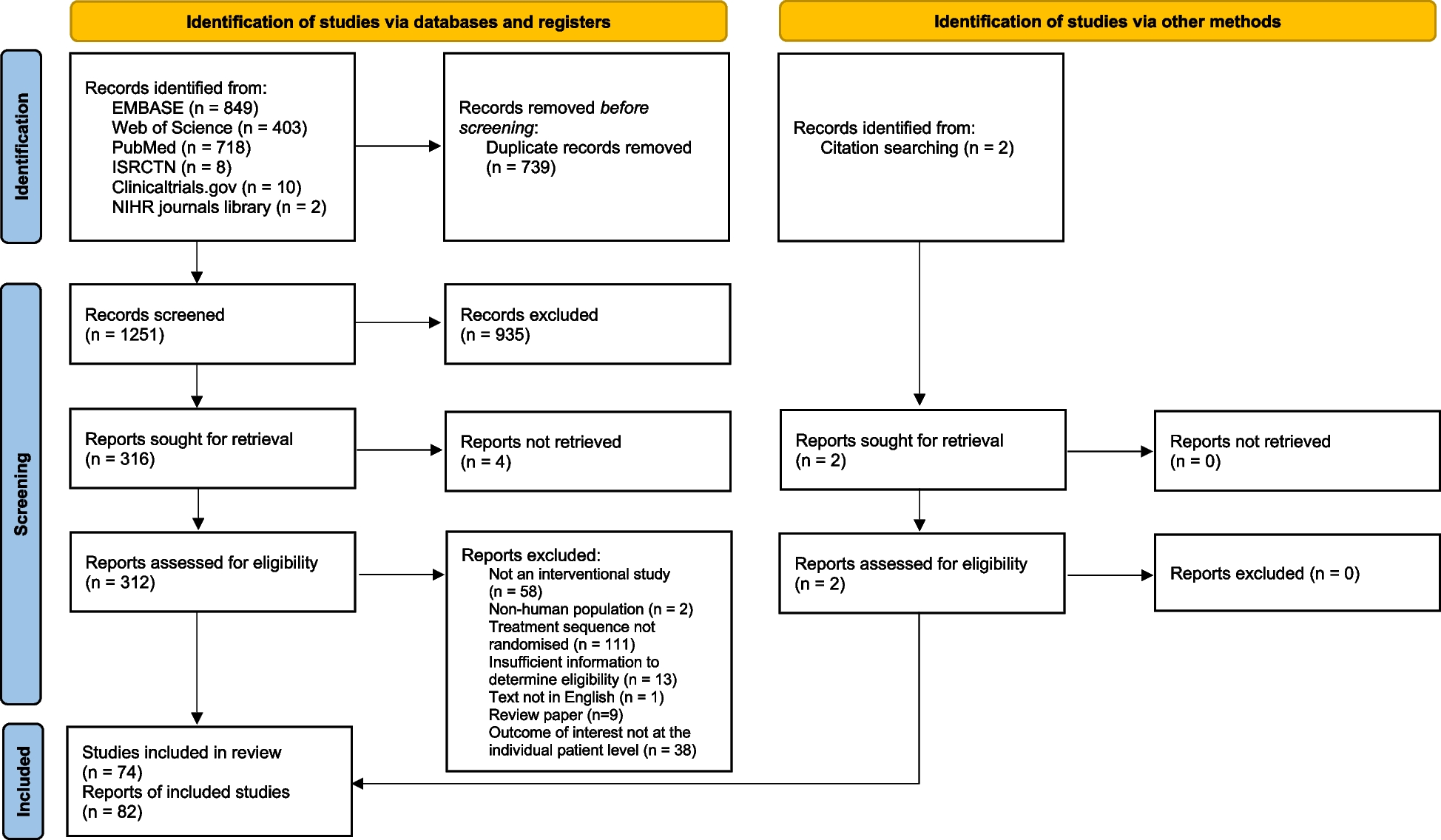
The participant timeline can be found in the study protocol [5]. A CONSORT diagram will be produced as part of the statistical analysis to show the flow of patients through the study, from recruitment through to treatment allocation, discharge, 30 days and 90 days follow-up.
Analysis populations
Each patient will be included or excluded from each of the analysis populations defined below. This will be carried out prior to unblinding to avoid bias.
Safety population
The safety population includes all subjects entered into the trial from the time of tracheal extubation up to 90 days after surgery (the period for which safety data is being collected) at the time of database lock. The safety population will be used to provide summary statistics on adverse events (AEs) and serious adverse events (SAEs), which will be reported by the treatment arm assigned during randomisation (or by the treatment arm actually received if it is different to that assigned during randomisation). The safety population will be examined at both the interim and final analyses. It will include patients with partial data as well as any patients who have withdrawn from the trial provided they continue to consent to their data being used.
Interim analysis population
The interim analysis population will include all patients who have been recruited at the time 300 patients have completed 90 days follow-up, including patients with partial data (e.g. anyone recruited after the 300th patient who has not yet completed their follow-up, and anyone recruited before the 300th patient who left the trial before completing follow up). If there is greater than 15% missing data for DAH90 (excluding missingness because of death, which is informative) and the interim analysis is delayed because of that, the interim analysis population may include more than 300 patients. The trial will recruit from the UK, Australia and New Zealand. It is expected that the majority of the patients recruited by the end of the trial will be from UK centres. To ensure that the results of the interim SSR reflect the expected proportions of UK, Australian and New Zealand recruits, we will seek to ensure that between 50 and 75% of the patients included in the interim analysis are recruited from the UK, with the remainder from Australia and New Zealand. This will ensure that even if differences exist between the countries, the effect will not disproportionately bias the interim SSR and therefore will not affect the power at the end of the trial. This requirement may impact on the timing of the interim analysis and may require that more than 300 patients have been randomised and followed up to 90 days to achieve the proportions stated.
Intention-to-treat population
The ITT population is the population that will be used for the majority of the analyses, including the analysis of the primary endpoint and the interim sample size re-estimation. The ITT population includes all subjects who were randomised, regardless of whether they received the treatment randomly allocated to them or completed follow-up. The data will be analysed assuming that the patient received the treatment they were randomly allocated to. If a patient dies after being randomised but before extubation occurs, they will be included in the ITT analysis population with a days at home score of zero.
Other populations
A per-protocol and two time-on-treatment (ToT) populations are also defined for use in sensitivity analyses. A full population is also defined. These are detailed in the additional document.
Protocol deviations and adherence
There are two analysis populations relating to protocol non-adherence. The ITT analysis population includes all subjects who were randomised, regardless of whether they received the treatment randomly allocated to them or completed follow-up. The data will be analysed assuming that the patient received the treatment they were randomly allocated. The per-protocol population includes all subjects who adhered to the trial protocol by receiving the treatment randomly allocated for a minimum of 16 h no matter whether they complete all of their follow-ups. Any subjects who did not receive the treatment randomly allocated to them will be excluded from the per-protocol population.
Descriptive statistics of compliance variables will be reported at the final analysis, split by treatment arm. These will include:
Summary of treatment compliance
Reasons for non-compliance
Summary of compliance for the ToT populations
Baseline characteristics
Baseline data will be collected following consent. This will include demographic data (age, sex, residential status, etc.), past medical history, quality of life (EQ-5D-5L), activity of daily living (BARTHEL), health service and resource use questions, the EuroSCORE II and the ARISCAT score. Descriptive statistics summarised by the treatment arm will be reported.
Interim analysis
The interim analysis will be performed after a minimum of 300 patients have been randomised and followed up for 90 days. If there is greater than 15% missing data for DAH90 (excluding missingness because of death, which is informative missingness) then the interim analysis may be delayed. If at the time 300 patients have been randomised and followed up, the rate of UK participation is outside the target range, the interim analysis may be delayed as per the definition of the interim analysis population.
Three main areas will be examined at the interim analysis:
The safety analyses at the interim analysis will be identical to the safety analyses at the final analysis, described below. Information on recruitment, treatment compliance, and data completeness will be reported at both the interim and final analysis. A summary of patient recruitment data will be presented by the centre, as well as by the treatment group and time or trial stage where appropriate. Compliance and data completeness will be summarised by the treatment group.
Sample size re-estimation
Due to uncertainty in the parameters used in the original sample size calculation and the sensitivity of the initial sample size calculation to changes in the nuisance parameters, NOTACS has been designed as an adaptive trial with an interim SSR planned after a minimum of 300 patients complete 90 days follow-up.
This sample size adaptation may prevent an underpowered trial if moderate deviations from the assumptions made for the initial sample size calculation are observed.
At the interim SSR, the accumulated data will be used to re-estimate a number of “nuisance” parameters including:
SD of DAH90 in the SOT arm
SD of DAH90 in the HFNT arm
Treatment switch rate from SOT to HFNT
Treatment switch rate from HFNT to SOT
Overall drop-out rate
Overall death rate
Treatment efficacy will not be assessed at the interim analysis.
An independent statistician will re-estimate the nuisance parameters using unblinded data (in order to allow the trial statisticians to remain blinded, and to preserve the type 1 error rate at 5%). The original sample size calculation will be repeated with the updated estimates to provide an updated sample size estimate which will be reported to the DMEC. The DMEC have the responsibility of agreeing the updated sample size following the rules in Table 1. Any recommendation outside of those listed in Table 1 should be clearly justified.
Table 1 Recommended sample size from interim SSR and course of actionNo adjustments will be made to the significance level due to the interim analysis.
Analysis for the primary endpoint
The analysis of the primary and secondary endpoints will take place at the end of the trial following database lock.
The primary outcome is DAH90. Following the approach of Myles et al., for the primary analysis, DAH90 will be treated as 0 for any patient that dies in the period between randomisation and the 90-day follow-up [9]. This definition is deemed appropriate on the basis that the death rate in the trial population is expected to be low (around 3%), most deaths are expected to occur within the initial hospital admission, the death rate is expected to be comparable between the two treatment arms, and it is not expected that the treatment will impact on the death rate. The primary analysis will be on the basis of intention to treat (ITT).
Due to the skewed nature of DAH scores, a Mann-Whitney-Wilcoxon test will be used for the primary efficacy analysis. Contrasts between treatment groups for DAH90 will be used to evaluate the difference in the median DAH90 at a 5% significant level. 95% confidence intervals giving a range of plausible effects will be reported. The non-parametric confidence interval will be calculated using either the Hodges-Lehmann method if an exact p-value is available, or a normal approximation otherwise.
The statistical analysis will be reported according to CONSORT extension guidelines for reporting of adaptive trials [10]. A review statistician will independently reproduce the final primary efficacy analysis.
Analysis for secondary endpoints
Important clinical covariates and sub-groups will be included in exploratory secondary analyses, and will include:
DAH90
DAH30
ARISCAT score
EURROSCORE II
Gender
COPD
Asthma
Obesity (BMI>35kg/m2)
Current smoking status
Lower respiratory tract infection in last 4 weeks
Age (≤ or > 80 years)
First time or re-do surgery
ROX index
Extubation timing (≤ or >24 h after admission to ICU)
Return to theatre (≤ or > 24 h of admission to ICU)
Length of initial ICU stay
Country
Centre (UK sites only)
Secondary outcomes
A series of Mann-Whitney-Wilcoxon tests will be used to assess whether a treatment effect exists for: DAH30; length of ICU stay during index admission; ROX index at 2, 6, 12, 24 and 48 h post-extubation; individual countries; and individual centres (UK only). Difference in medians with corresponding 95% confidence intervals will be reported for each comparison. The non-parametric confidence interval will be calculated using either the Hodges-Lehmann method if an exact p-value is available, or normal approximation otherwise.
Chi-squared tests (or Fisher’s exact test where appropriate) will be used to assess the association between the treatment group with extubation timing (≤ or >24 h after admission to ICU) and return to theatre (≤ or > 24 h of admission to ICU).
To allow for the estimation of effect sizes and adjustment for potential clinically important covariates, quantile regression models will be fitted for DAH90 and DAH30 as outlined in Table 2. Quantile regression was chosen due to the heavily skewed nature of the DAH endpoints and due to the expectation of two spikes close to day 0 and day 90/30.
Table 2 Quantile regression modelsSensitivity analyses
A number of sensitivity analyses will be performed to examine the robustness of the primary analysis. These include:
Using the per-protocol population instead of the ITT population;
Using each of the two ToT populations instead of the ITT population;
Using an alternative definition of DAH90 for patients that die during follow-up;
Examination of missing data, with multiple imputation of missing data where appropriate.
Further details of these sensitivity analyses can be found in the additional document.
Safety analyses
All safety analyses will be performed on the safety population. Data on AEs and SAEs will be collected from the time of tracheal extubation to discharge. From discharge up to 90 days after surgery, only data on SAEs will be collected. Adverse events (AEs) and serious adverse events (SAEs) will be summarised separately. AE and SAE data will be listed by MedDRA Preferred Term and further grouped by System Organ Class. The frequencies of the AEs will be summarised by treatment groups. Death, stroke, sepsis, myocardial infarction and acute kidney injury are SAEs of special interest. Therefore, the rates of these will be estimated for each treatment group separately to the main SAE summary table.
Other analyses
Descriptive statistics and summaries of recruitment and compliance will be examined at the final analysis (see additional document for details).




留言 (0)