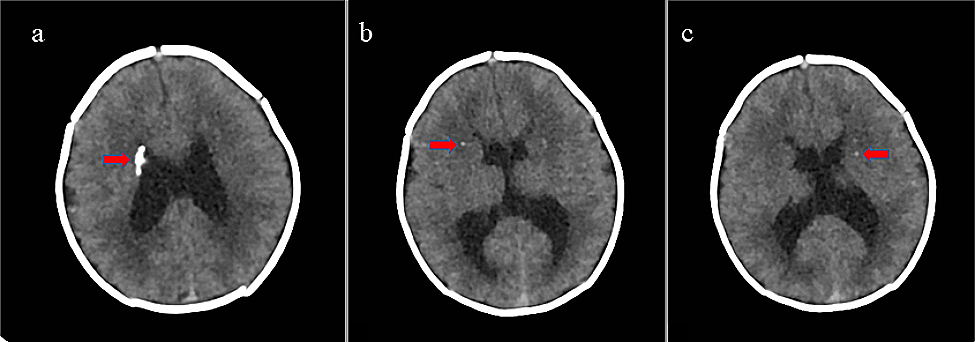
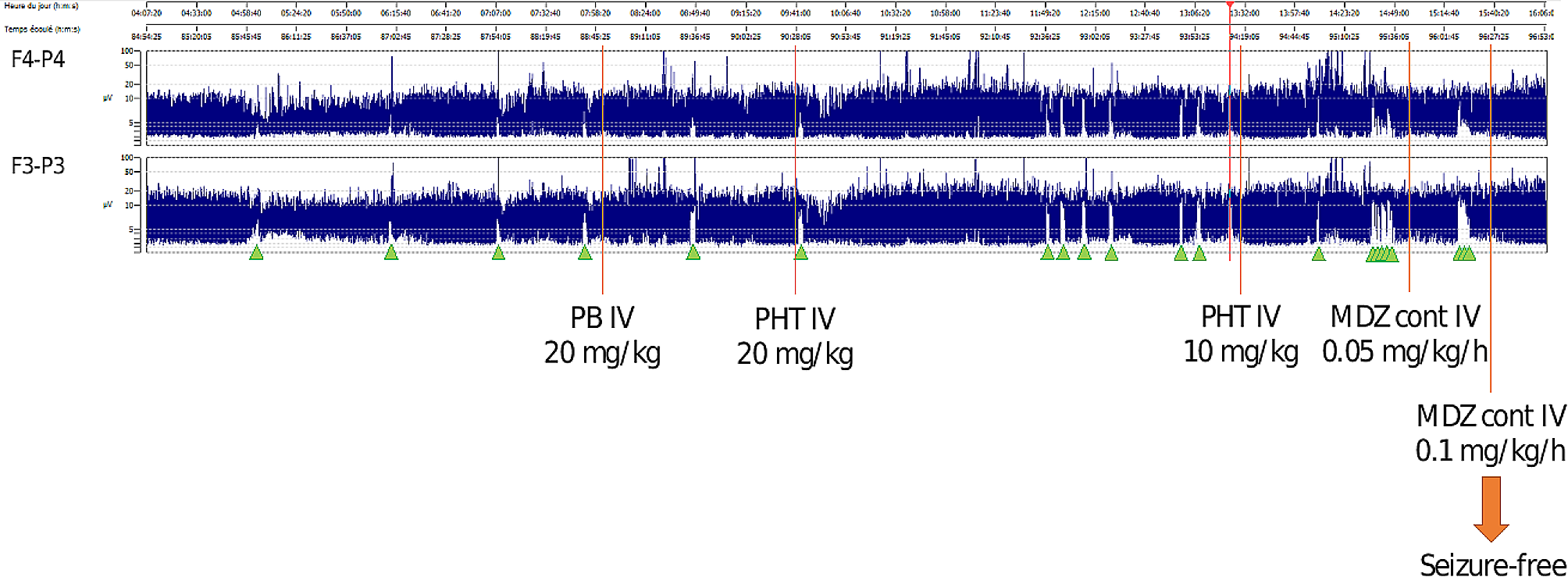
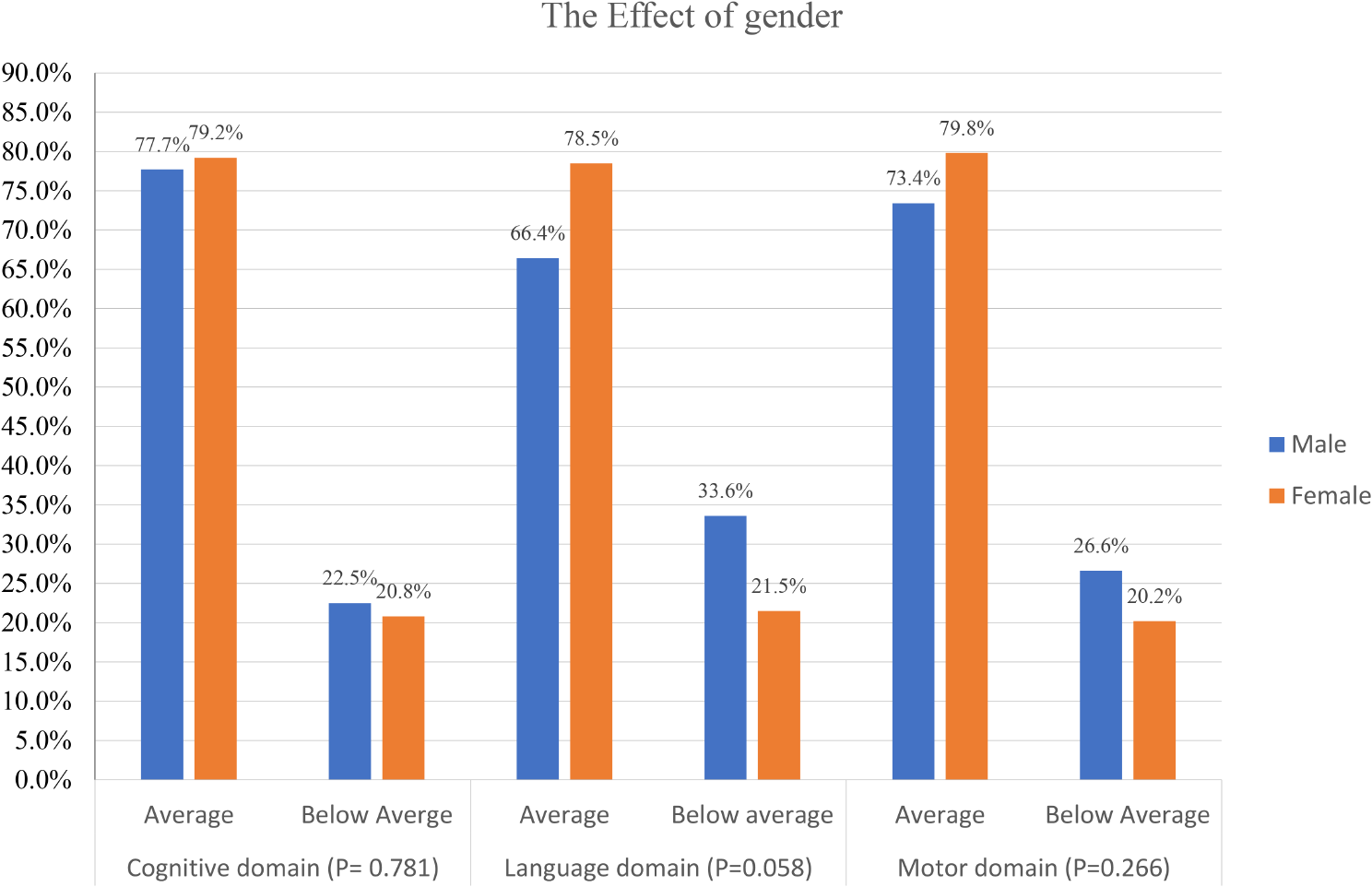
Noonan and Noonan-like syndromes are multisystem genetic disorders, mainly with autosomal dominant trasmission, caused by mutations in several genes: PTPN11 (OMIM 176,876), KRAS (OMIM 190,070), SOS1 (OMIM 182,530), RAF1 (OMIM 164,760), NRAS (OMIM 164,790), BRAF (OMIM 164,757), RIT1 (OMIM 609,591), SOS2 (OMIM 601,247), LZTR1 (OMIM 600,574), MRAS (OMIM 608,435), RRAS2 (OMIM 165,090), MAPK1 (OMIM 176,948), RRAS2 (OMIM 600,098), A2ML1 (OMIM 616,027), CBL (OMIM 165,360), SHOC2 (OMIM 602,775), PPP1CB (OMIM 600,590).
The PTPN11, SOS1, KRAS, RAF1, BRAF and MEK1(MAP2K1) genes, account for approximately 70% of affected individuals. SHP2 (encoded by PTPN11), SOS1, BRAF, RAF1 and MEK1 positively contribute to RAS-MAPK signaling through complex autoinhibitory mechanisms, that fail when these genes have mutated.
Whatever the altered gene is, the expressiveness of the clinical picture is variable even within the same family. Clinically NS is characterized by the presence of postnatal growth impairment with short stature, peculiar craniofacial characteristics and congenital heart disease as pulmonary valve stenosis (50–60%), hypertrophic cardiomyopathy (20%), atrial septal defects (8%), interventricular septal defects (5%). Electrocardiographic changes including left axial deviation, an abnormal R / S ratio on the left precordial leads and an abnormal Q wave, or heart rhythm disturbances in patients with hypertrophic cardiomyopathy are reported in a variable percentage of patients. Other signs frequently associated with the syndrome are pterygium colli, deformity of the rib cage, delayed psychomotor development of varying degrees, anomalies of the genitourinary system (cryptorchidism, hypospadias, micropenis), anomalies of the lymphatic system (hypoplasia of the lymphatic vessels), ocular abnormalities (strabismus, myopia), ectodermal component and dentition anomalies (pilar keratosis, skin discoloration, bristly or fluffy hair, malocclusion) and bleeding tendency for coagulation anomalies with deficiency of some coagulation factors. An increased susceptibility to develop autoimmune diseases (celiac disease, vasculitis, thyroiditis, SLE, uveitis) and myeloproliferative disorders have also been reported. [1, 36, 38, 40]
In addition to myeloproliferative disorders and acute lymphoblastic leukaemia, several solid tumors have been reported in individuals with NS, mainly embryonal rhabdomyosarcoma, neuroblastoma, and glial tumors.
Although the tumor risk in patients with related SOS1 NS was previously considered lower than in other forms linked to other genes, over the years a significant incidence of some solid tumors has been reported in these patients including embryonal rhabdomyosarcoma, Sertoli cell testis tumor, granular cell tumors of the skin and mandibular multiple giant cell lesions (MGCLs). These are benign tumor-like lesions consisting of an osteoblast-like cell population, already described in patients with NS or other RAS-opathies, whose higher incidence has been described to be linked to cases due to mutations in SOS1 [2, 7, 13, 16, 20, 24, 25, 28, 35].
The dermatological findings of RASopathies consist in pigmented lesions (café-au-lait spots, lentigines, and melanocytic lesions), ectodermal lesions (ichthyosiform manifestations, follicular hyperkeratosis, short, curly, thin hair), and hyperplasia (redundant skin, papillomatous growths). Several of these skin manifestations are associated with a specific syndromic phenotype, even if the molecular mechanism determining the association of specific tegumentary lesions with specific syndromes and specific genes is still unknown.
More in detail, the abnormalities observed in patients with NS appear to depend on the mutation responsible, indeed it was observed that hyperkeratotic skin is much more frequent in NS patients harboring SOS1 gene mutations and generally in subjects with mutations in genes directly involved in cell proliferation kinase cascades (SOS1, BRAF, KRAS and RAF1) [3, 14, 30, 31, 37].
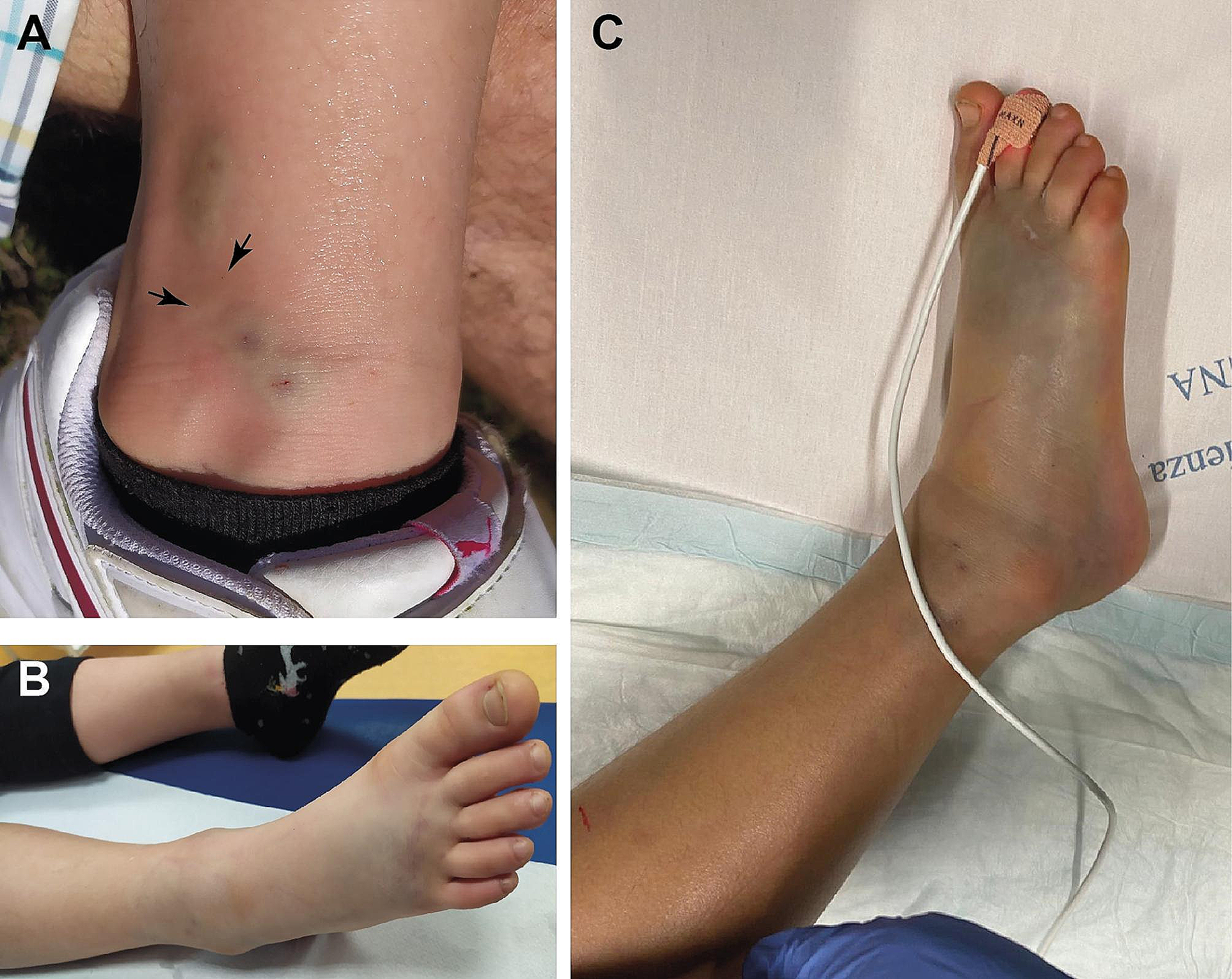
Pathogenetic variants of SOS1 are responsible for NS but also for hereditary gingival fibromatosis type 1, a benign overgrowth condition of the gingiva characterized by a slowly progressive, benign fibrous enlargement of keratinized gingiva. Missense mutations of this gene, as in the cases under examination, are the second most common cause of NS and account approximately for 13% to 17% of cases. Subjects carrying a pathogenetic variant in SOS1 gene tend to exhibit a distinctive phenotype that is characterized by ectodermal abnormalities (keratosis, pilaris, hyperkeratocic skin, sparse eyebrows, sparse thin and curly scalp hairs) generally not associated with cognitive deficits in place of which mood disorders such as anxiety or depression have been described along with absence of growth impairment. In some subjects, fetal macrosomia, found in the prenatal period, was not correlated with postnatal growth. Patients with SOS1-related NS display typical facial features, including macrocephaly, hypertelorism, ptosis, downslanting palpebral fissures, sparse eyebrows with keratosis pylaris, curly hair a short and broad nose with upturned tip, low-set and posteriorly angulated ears, and high forehead commonly associated with bitemporal narrowing and prominent supraorbital ridges. Among the heart defects in these subjects pulmonary stenosis, atrial and ventricular septal defects, prevail, while hypertrophic cardiomyopathy is rarer (almost 10%). Digestive disorders such as gastroesophageal reflux disease, Chiari malformation, refractive disorders and musculoskeletal pain are also reported in adolescent and / or adult subjects [5, 14, 15, 24, 27, 31, 37, 42].
SOS1 gene (OMIM 182,530) encodes the Ras-GEF protein SOS1 (SOS RAS/RAC guanine nucleotide exchange factor 1), which, once translocated on the plasma membrane, binds Ras stimulating its conversion from the inactive GDP-bound form to the active GTP-bound form [11, 24, 39].
It is a large multidomain protein characterized by an N-terminal regulatory portion including tandem histone-like folds (HF), which are followed by a Dbl-homology (DH) domain, a pleckstrin-homology (PH) domain, and a C-terminal catalytic region including the RAS exchanger motif (REM) and CDC25 domains, followed by a tail providing docking sites for adaptor proteins required for receptor anchoring. The ordinary conformation of DH and PH domains (the DH-PH unit) usually blocks allosteric Ras binding. HF also performs this function and besides stabilizes the autoinhibitory conformation of the DH-PH unit. Moreover, both HF domain and the DH-PH unit are conformationally coupled to control SOS1's recruitment to the plasma membrane. This process is a necessary step to alleviate the effect of the HF and DH domains and allow the Ras link to the allosteric site that promotes a conformational rearrangement of the CDC25 domain promoting RAS binding to the catalytic site. SOS1 disease causing variants are almost always missense changes, most of whom are found in exons coding domains HF, DH and PH. Since these stabilize SOS1 inhibited conformation, their disruption causes a release of autoinhibition and consequently a gain-of-function of SOS1 that involves an increase in the active form of Ras and greater Ras/MAPK pathway signaling [11, 24, 38, 39, 42].







留言 (0)