
記住我
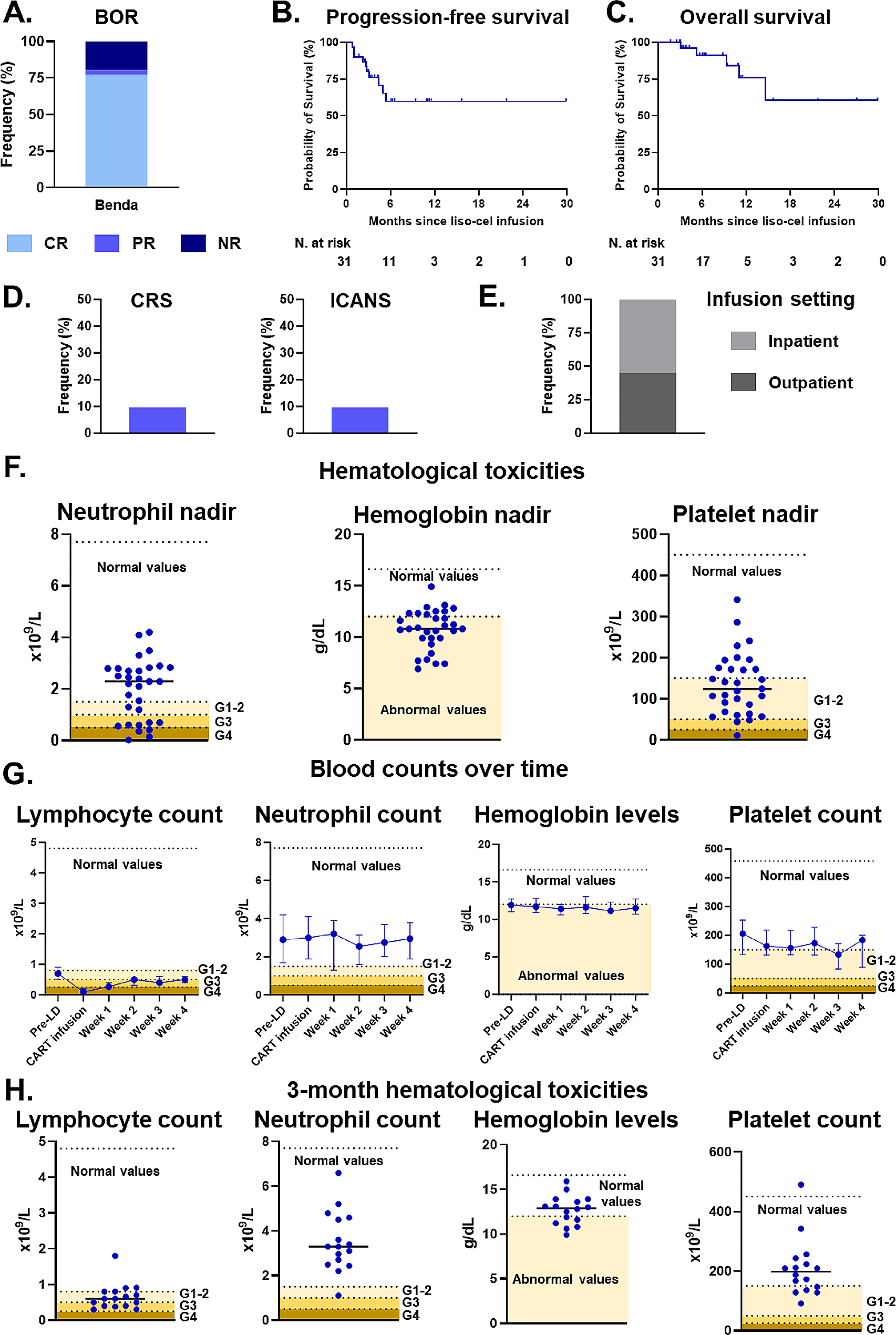



A 61-year-old male with penta-refractory MM (IgA lambda), ISS stage 3 with hyperdiploidy, gain of 1q21 and del13 was treated with anti-BCMA CAR-T cell therapy, achieving a best response of VGPR. He progressed after 6 months and was temporarily salvaged with BCNU/Melphalan (carmustine 300 mg/m2 and melphalan 140 mg/m2) as conditioning regimen before ASCT, achieving a best response of PR until further progression with extramedullary disease (subcutaneous skin lesions in lower extremities) and elevated lambda free light chains (FLC, 126.4 mg/l) at 6 months (Fig. 1) (Additional file 1).
Fig. 1
Timeline depicting the patient’s clonal evolution, treatment regimens, and responses since diagnosis. Timeline is represented in days since the establishment of MM diagnosis. Treatment regimens are indicated with black arrows descending toward the upper side of the timeline. Diagnostic whole-exome sequencing (WES), RNA-sequencing (RNA-seq) or targeted panel are indicated as blue bars descending from the timeline. Red arrows indicate period of disease progression (PD), and different shades of green arrows indicate very good partial response (VGPR), complete response (CR), and stringent complete response (sCR). Lower part of the graph depicts the patient’s lambda LC levels (mg/L) since the beginning of the triple therapy regimen (regorafenib, dabrafenib and encorafenib) until sCR achieved after antibody trial 2. ASCT: Autologous stem cell transplantation; VCd: Velcade (Bortezomib), cyclophosphamide and dexamethasone. VRd: Velcade (Bortezomib), lenalidomide, and dexamethasone; Vd: Velcade (Bortezomib) and dexamethasone; anti-BCMA CAR-T: anti-B cell maturation antigen chimeric antigen receptor T cells; VDCEP: Velcade (Bortezomib), dexamethasone, cyclophosphamide, etoposide, and cisplatin; BCNU: Carmustine
Prior to CAR-T therapy, targeted and WES had identified a BRAF(V600E) mutation in his BM, which persisted throughout treatment and was also detected in a cutaneous plasmacytoma with a variant allele frequency (VAF) of 41% at relapse from CAR-T. Western blot (WB) analysis of the post-CAR-T relapse bone marrow aspirate (BMA) confirmed the BRAF(V600E) mutation and further showed phosphorylation of ERK (pERK), consistent with activation of MAPK signaling, which was inhibited by combining the MEK inhibitor binimetinib with the BRAF(V600E) monomer inhibitor encorafenib. Imaging the blot again with longer exposure time revealed small residual MAPK activity, which was abrogated by adding the BRAF dimer inhibitor regorafenib (Fig. 2A and C). Further assessment of ex vivo drug sensitivity of the patient’s CD138+ MM cells post-CAR-T to different combinations of MAPK inhibitors through a functional assay based on measurements of single-cell mass response suggested high efficacy of the combination of trametinib at 5 µM and dabrafenib at 1 µM. Adding regorafenib at 1 µM indicated improved response compared to the double combination (Fig. 2B) [6]. According to our in vitro experimental results and previous literature on MAPK pathway homeostasis, phosphorylated ERK—the downstream effector of activated MAPK pathway—exerts a negative feedback inhibition on the receptor tyrosine Kinase (RTK). When pERK levels decrease upon RAF monomer inhibition ± MEK inhibition (standard approach), this negative feedback is eliminated, i.e., Relieve of negative feedback, resulting in RTK and subsequent MAPK activation, which is known as feedback recovery. This is mostly mediated by the formation of dimeric RAF. Therefore, adding a dimer selective RAF inhibitor, i.e., regorafenib, overcomes this feedback recovery [7,8,9].
Fig. 2
Triple MAPK inhibition effectively reduces phosphorylated ERK in BRAF (V600E) CD138+ plasma cells A WB of magnetic bead selected CD138+ plasma cells from RRMM patient’s BMA after 48 h in vitro treatment with encorafenib (ENC; 50 nM) and binimetinib (BIN; 250 nM), regorafenib alone (REG; 1μΜ), or combination of the three drugs. B Travera analysis on RRMM patient CD138 + cells showing sensitivity to trametinib (TRAM) in combination with dabrafenib (DAB) and regorafinib (REG) at varying concentrations. C Relative pERK protein expression after quantification and normalization to actin
Based on these findings and assessment, the patient was started on targeted therapy with a combination of a BRAF monomer inhibitor, dabrafenib (100 mg, orally twice daily), a MEK inhibitor, trametinib (1.5 mg, orally for 21/28 days daily), and a BRAF dimer inhibitor, regorafenib (40 mg, orally once daily). Within 3 months of treatment initiation, prompt reduction in subcutaneous skin lesions and 80% reduction in Lambda free light chain (λFLC) (27.5 mg/l) were observed (Fig. 1). Furthermore, the patient had good tolerance to all three medications with minimal side effects (grade 1 fatigue) which allowed him to carry out activities of daily living and return to work.
After more than three months of optimal treatment response, the patient’s FLC started to gradually increase, indicating reduced response to the triple MAPK inhibition, albeit with no recurrence of the subcutaneous nodules. The triple MAPK inhibition dose was maximized to dabrafenib 150 mg (orally twice daily), trametinib 2 mg (orally once daily), and regorafenib 80 mg (orally once daily), for less than a month, and eventually discontinued when no further response was observed with FLC peaking at 116.6 mg/l (Fig. 1). The 110-day response period achieved by the triple MAPK inhibition allowed the patient to become eligible for clinical trials and is currently benefiting from bispecific treatment with a λFLC of 10.9 mg/l with FLC ratio of 0.49 indicating stringent complete response (sCR).
To better understand the evolutionary trajectory of the alterations in the MAPK pathway over time, we analyzed matched DNA and RNA-sequencing from CD138+ samples taken at various time points from prior to CAR-T therapy to after relapse from triple MAPK inhibition. The BRAF V600E mutation was subclonal, yet detected relatively early within the tumor clonal phylogeny, and was present in the bone marrow prior to CAR-T therapy (VAF = 12.8%, Fig. 3A–B). During CAR-T therapy, the subclone harboring this mutation reduced in cellular fraction to undetectable levels, then recovered upon the patient’s relapse of CAR-T in the cutaneous plasmacytoma (VAF = 40.7%) and was finally detectable in the bone marrow prior to the start of triple MAPK inhibition (Fig. 3A). The BRAF V600E subclone further expanded at relapse from triple MAPK inhibition, producing a descendant subclone harboring a KRAS Q61R mutation (Fig. 3B). The new KRAS Q61R mutated subclone was detectable in trace proportions immediately after relapse from triple MAPK inhibition (VAF = 2.7%) and expanded further 30 days later (VAF = 14.4%, Fig. 3A). Longitudinal pathway analysis in RNA-seq data also revealed a successive upregulation of MAPK and PI3K pathway activity after relapse from CAR-T and throughout the course of MAPK inhibition (Figs. 3C and 4D).
Fig. 3
Temporal evolution and trajectory of MAPK alterations. A Clusters of subclonal mutations sampled over time with the mutational VAF represented as a percentage on the y axis show the trajectory of the subclone harboring the BRAF V600E mutation over time in response to treatment and the later emergence of a subclonal KRAS Q61R mutation after the end of triple MAPK inhibition. B A reconstructed phylogenetic tree of subclones across all time points shows that the BRAF V600E is subclonal and ancestral to the clone that gives rise to a KRAS Q61R mutation. C RNA expression shows an increase in MAPK pathway activation, measured as a combined z-score, over time in response to treatment with triple MAPK inhibition therapy
Fig. 4
CD138+ BMA plasma cells shift their dependency to PI3K/AKT pathway with increased sensitivity to copanlisib A WB of magnetic bead selected CD138 + plasma cells from RRMM patient’s BMA after 48 h in vitro treatment with with encorafenib (ENC; 50 nM) and binimetinib (BIN; 250 nM), regorafenib alone (REG; 1μΜ), combination of the three drugs or copanlisib alone (25 nM). B Rlative protein expression of pERK, pS6, pAKT and BRAF (V600E) after quantification and normalization to actin C CD138 + viability measurement after 48 h in vitro treatment with with encorafenib (ENC; 50 nM) and binimetinib (BIN; 250 nM), regorafenib alone (REG; 1 μΜ), combination of the three drugs or copanlisib alone (25 nM). D RNA expression shows elevated PI3K/Akt pathway activation, measured as a combined z-score, throughout the course of treatment with triple MAPK inhibition therapy
Post-MAPK inhibition western blot analysis revealed only slight reduction in phosphorylated ribosomal S6 upon ex vivo triple MAPK therapy, despite effective inhibition of the MAPK pathway with undetectable level of phosphorylated ERK. This suggests that the patient’s plasma cell clones might have shifted their dependency to an alternative pathway, as suggested by increased PI3K signaling transcriptional output, based on RNAseq analysis. In fact, treatment of the MM cells with the PI3K inhibitor copanlisib could effectively inhibit phosphorylated ribosomal protein S6, indicating higher clonal dependency on PI3K (Fig. 4A and B). Pathway dependency was further evaluated by measuring cell death in a 48 h in vitro assay with patient’s selected CD138+ cells with different drug combination. Triple therapy achieved 10% cell death while copanlisib approximately doubled cell death of the plasma cell subclones (Fig. 4C). This indicates that eventual failure of triple MAPKi therapy may be a result of tumor clonal evolution, as well as a shift in cell dependency toward PI3K pathway activation.
留言 (0)