
記住我










The COVID-19 pandemic has led to an emergency scenario due to the virulence and contagiousness of the virus and to its rapidity in causing a high percentage of serious clinical conditions, worsened by the absence of specific drugs.
Antiretroviral drugs were among the first medicines used in COVID-19 disease, both in early stages and in advanced intensive care,1 2 for which clinical trials are ongoing to verify the benefits from treatment.3–9 The lopinavir/ritonavir combination, licensed in the treatment of HIV, is marketed in Italy only in tablets. Liquid formulation for oral administration is available on the European market as Kaletra (80 mg+20 mg/mL) solution, stored in refrigerated conditions, but is not promptly accessible in Italy, due to the authorisation needed for importation and, consequently, delivery times. In COVID-19 cases, great effort has been made in developing suitable therapeutic formulations for administration to passive patients (sedated and intubated) as in the cases requiring an alteration of dosage form.10
Parenteral administration is the best choice for critical patients, but the absence of injectable formulations necessitates the administration of liquid drugs through a nasogastric tube (oral solutions, suspensions or syrups).
Magistral formula preparation by the hospital pharmacy as a galenic oral formulation from the tablets represents a useful alternative for patients who require a specific route of drug administration. It can also be useful in other contexts where the supply chain is critical or the management of refrigerated medicines constitutes a structural limitation, necessitating the occasional use of crushed tablets.
Based on the Good Drug Preparation Rules, defined by the Italian Pharmacopoeia, a magistral formula preparation must guarantee quality, safety and efficacy. Various aspects must be assessed on which those criteria depend. First, a formulation setup, including simple manipulations in the pharmacy and the ward, is recommended above all for ready-to-use forms. A second aspect that must be evaluated is the primary packaging of the formulation to prevent the clinical risk associated with the wrong administration route.11 The stability of the formulation is another important aspect to ensure therapeutic continuity in the time slots in which the pharmacy is not able to compound galenic formulations.
Furthermore, for the galenic formula it is also of primary importance to evaluate the precise content of the active pharmaceutical ingredient (API) in the new pharmaceutical form and how it is directly influenced by the solubility in the main solvent, usually water. Indeed, lopinavir and ritonavir are practically insoluble in water,12 but soluble in alcohols. It can be consistently observed that the commercial liquid oral formulation has an ethanol content of 42.4%.13
A preliminary study of the composition of the commercial formulations of lopinavir/ritonavir was undertaken to list the potentially useful excipients for formulating an oral liquid galenic preparation with the best availability of the APIs comparable to that of a marketed liquid formulation.13
We describe here a new galenic formula of lopinavir/ritonavir (figure 1) prepared from crushed tablets in water, by using ethanol and glycerol as co-solvents by virtue of their physicochemical characteristics. In order to identify the optimal formulation procedure, several issues related to pharmaceutical quality must be addressed, such as the exact amount of dissolved APIs, possible contaminations due to impurities from solvents, as well as, importantly, the chemical stability over time of the APIs, which imposes constraints to shelf life. The use of several analytical techniques could be required to deal with all the above aspects, the most common of which, however, are based on the comparison with suitable standards having known structure. On the other hand, high resolution 1H nuclear magnetic resonance (NMR) spectroscopy represents a powerful investigation tool which allows us to observe, simultaneously by a single measurement, the different kinds of hydrogen (1H) nuclei in every component of the mixture: APIs, solvent, co-solvents and impurities arising from degradative processes. Importantly, the comparison of the integrated areas of the 1H signals is exclusively affected by component percentages. By selecting an external standard compound with known concentration, the absolute amounts of components can be reliably and carefully determined.14–17 No manipulation of the samples is required, since they are simply transferred into the NMR tube before analysis.
 Figure 1
Figure 1 Chemical structures of lopinavir and ritonavir.
Although high resolution NMR spectrometers are not commonly available in hospital contexts, collaboration with different kinds of scientific institutions nowadays make their use easily accessible. Working teams with specific skills can be organised, able to answer in real time questions about the stability and safety of galenic preparations, to ensure the quality of APIs and consequently of galenic productions, in timing consistent with healthcare activities in emergency situations.
Materials and methodsMaterialsCommercial products: lopinavir 200 mg/ritonavir 50 mg (Mylan SpA) Kaletra oral solution (AbbVie Deutschland GmbH & Co KG), ethanol 96% Ph.Eur, (Carlo Erba Reagents Srl), glycerol ≥98% Ph.Eur (Olcelli Farmaceutici Srl); water highly purified (SALF Laboratorio Farmacologico); CD3OD (Deutero GmbH); and enteral syringe IFU Nutrifit (Vigon, Pentaferte).
Comparative analysis of commercial drug formsTo obtain a reproducible and ready to use galenic formulation starting from any commercial tablet form of lopinavir/ritonavir, the composition of the commercial products on the Italian and foreign market were compared: Kaletra tablets; lopinavir/ritonavir Mylan tablets; lopinavir/ritonavir Accord tablets; Aluvia tablets. They have a similar composition both for the tablet core and the protective film.13 Qualitative and quantitative Kaletra oral solution composition was assessed too. Marketed oral solution contains ethanol (42.4% v/v), propylene glycol (15.3% p/v) and high fructose corn syrup (168.6 mg/mL) as the main excipients.
Galenic formulationsTo define the best excipient proportion and preparation method, three different formulations were studied, using ethanol and glycerol as excipients, and two different preparation procedures (two-step vs one-step). The ethanol content in a single dose of the galenic formulation (total volume about 27 mL) and in the 5 mL dose of Kaletra oral solution is the same. The influence of the coating film removal on the dispersing ability and API content was investigated. Results have been compared with Kaletra oral solution and simple water dispersion of crushed tablets.
Preparation method A (two-step)Two lopinavir 200 mg/ritonavir 50 mg tablets were used for each dose. Both tablets were crushed in a mortar to fine amorphous powder, which was collected into the screw cap container. Ethanol/glycerol solution was added to the powder and shaken by dynamic horizontal movements for 10 s to completely wet the powder, and then it was left to rest for 10 min. Water was added and the mixture was shaken vigorously to obtain a homogeneous suspension.
Preparation method B (one-step)Two lopinavir 200 mg/ritonavir 50 mg tablets were used for each dose. Both tablets were crushed in a mortar to fine amorphous powder, which was collected into the screw cap container. Glycerol-hydroalcoholic solution, in a single phase, was added and shaken by dynamic horizontal movements for 10 s to facilitate powder dispersion, and then it was left to rest for 10 min before administration. In table 1 galenic liquid formulations according to methods A and B are shown.
Table 1Galenic liquid formulations: composition of the dispersing phases for two crushed tablets; (glycerol) is the glycerol concentration in the final formulation
API solubilisation tests were done on crushed tablets both with and without coating film (A2 vs A4, table 1). The action of removing film needs manual skill to avoid losing the product, and therefore the weight was checked before and after, to verify removal efficiency and to assess removal effects on API content in the final dispersion. The effect of removing coating film was studied for the intermediate concentration of glycerol (11%). Regarding preparation method A three different dispersing phases were tested with glycerol concentration of 6%, 11% and 15% (A1–A3, table 1). Regarding preparation method B, a single hydroalcoholic dispersing phase with 11% glycerol or only water was analysed (B1–B2, table 1). Stability of all the formulations was studied over 48 hours at 4°C.
Table 2 summarises the composition of binary ethanol/glycerol mixture and the content of ethanol and glycerol in the final formulation. Ethanol concentration in the final formulation was kept constant except for entry 6-A1.
Table 2Galenic liquid formula: preparation and composition (v/v %) of phase 1 (ethanol/glycerol) and of the final formulation
NMR instrumentationNMR measurements were performed on a Varian INOVA600 spectrometer (Varian Inc, Palo Alto, CA, USA) operating at 600 MHz for 1H. The temperature was controlled to 25±0.1°C. Quantitative 1H NMR spectra were acquired by using a relaxation delay of 10 s, and an observation pulse corresponding to 60°. In all cases the same experimental acquisition parameters were used to guarantee accurate quantitative analysis.
Samples for NMR analysesAll of the NMR samples were prepared by mixing an aliquot (usually 0.2 mL) of the oral formulations A1–B2 of table 1 directly into the NMR tube with an aliquot (usually 0.5 mL) of the deuterated solvent (CD3OD), which is needed for the common lock and shimming procedures. Afterwards, the NMR samples were stored at 2–4°C.
The commercially available oral formulation Kaletra (lopinavir 80 mg/mL, ritonavir 20 mg/mL) was analysed in the same experimental conditions as the galenic oral formulations obtained from the solid form of lopinavir 200 mg/ritonavir 50 mg Mylan tablets. The NMR sample of Kaletra (0.1 mL of Kaletra in 0.6 mL of CD3OD), containing lopinavir 8 mg/mL and ritonavir 2 mg/mL, was used as the external standard for quantitative analysis.
Results and discussionAmong the available excipients with good solubilising, rheological and preservative properties, glycerol is an excellent additive because of its wetting, thickening, lubricating and preservative features. Ethanol also solubilises APIs contributing to preserving the liquid formulation.18 Liquid galenic formula of lopinavir/ritonavir compositions and preparation methods were tested using different ethanol/glycerol dispersing phases (entries 2–6, table 2).
The quantitative NMR analyses of such formulations were carried out directly on diluted formulations. APIs were quantified by diluting a known amount of each formulation with a constant volume of CD3OD and by recording quantitative NMR spectra. The marketed oral formulation Kaletra was used as the external standard.
The proton NMR spectrum of the oral formulation Kaletra is shown in online supplemental figure S1. Among NMR resonances produced by APIs in the spectrum, the signal of the aromatic proton of the ritonavir thiazole moiety at 8.94 ppm was chosen for the quantitative determinations. This signal occurs in a spectral region free of excipient signals and also for the galenic formulations obtained from crushed tablets. As the lopinavir/ritonavir ratio was found to be constant in all the samples, the determination of ritonavir allows the simultaneous determination of liponavir.
The galenic oral formulations, prepared in two steps (A-type) or in one step (B-type), were analysed by NMR and the concentration of the APIs was determined 30 min (t0) and 48 hours (stored at 4°C) after the preparation to monitor the content of antiretroviral drugs in these dosage forms and their physicochemical stability.
Figure 2 compares the NMR spectra of the different galenic formulations, and the results of quantitative analyses are summarised in table 3.
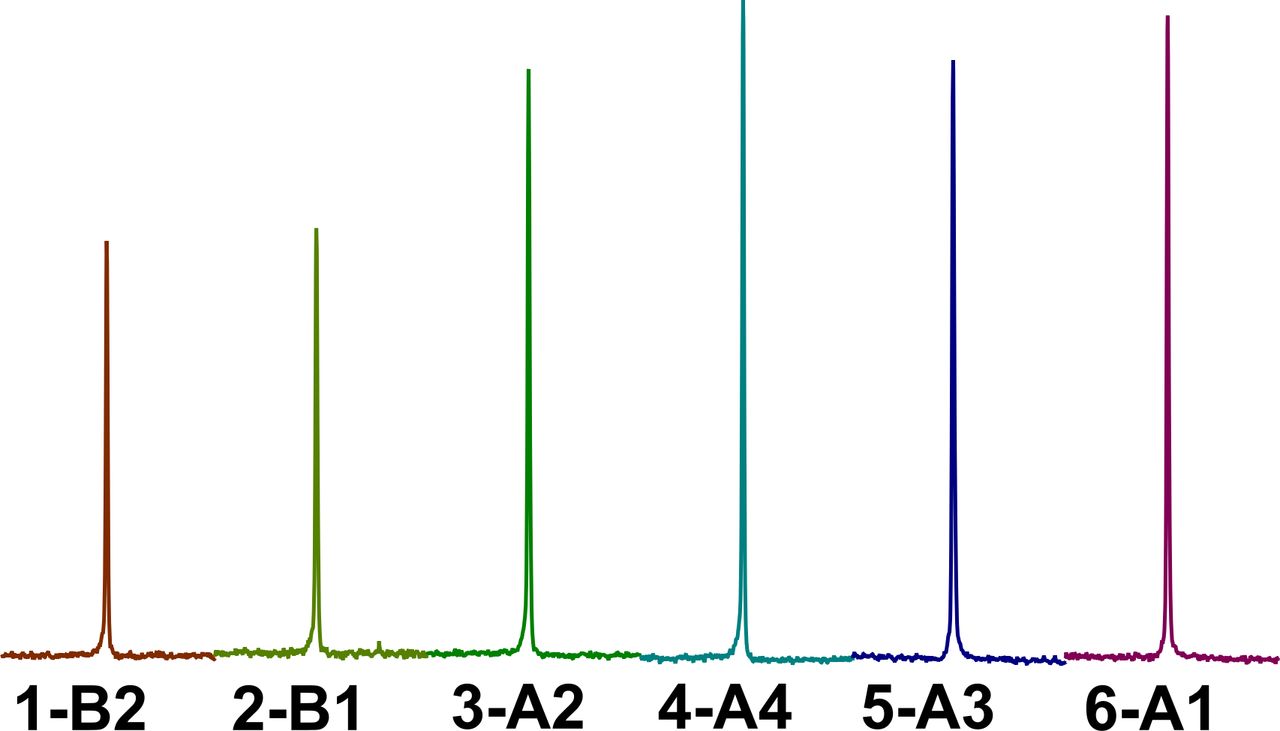 Figure 2
Figure 2 1H NMR (600 MHz, CD3OD, 25°C) spectral regions corresponding to the selected aromatic proton of ritonavir in the six galenic formulations. NMR, nuclear magnetic resonance.
Table 3Quantitative NMR analysis of API content in galenic liquid formulations 30 min (t0) and 48 hours at 4°C after preparation
In the preparation entry 1-B2 (table 2), involving simple dispersion of the crushed tablets in water, without co-solvents, the content of the active ingredients decreased notably by about 40% compared with the commercial syrup (table 3). The same preparation also showed poor stability, since the concentration of the active ingredients decreased from 63% at t0 to 56% after 48 hours stored at 4°C (table 3).
The presence of ethanol and glycerol as co-solvents is crucial both to improve solubilisation and promote the stability of its oral form, as long as a two-step method (preparation method A) is followed in the preparation procedure. In fact, when crushed tablets were first dispersed in the ethanol/glycerol mixture and then in water (entries 3-A2, entry 4-A4, entry 5-A3, and entry 6-A1, table 3), the content of solubilised active ingredients was equal to or only slightly lower than the standard Kaletra, ranging from 89% to 100%. By contrast, when the crushed tablets were dispersed in one step (preparation method B) in the solvent mixture simultaneously containing ethanol, glycerol and water (entry 2-B1, table 3), the amount of APIs was comparable (65%) to that obtained by using water as the sole dispersing medium (entry 1-B2, 63%, table 3). Increasing glycerol content in the formulations did not seem to significantly affect the drugs’ dissolution (entry 3-A2 and entry 5-A3, table 3), but it may have produced a relevant increase in the formulations’ viscosity. Ethanol is certainly the component in the co-solvent mixture contributing more to the solubilisation processes of the lipophilic active ingredients, as demonstrated by comparing quantitative data of entries 6-A1 (97%), 3-A2 (89%) and 5-A3 (91%) (table 3), where 55%, 40% and 32% of ethanol in phase 1 were respectively present. In particular, in the presence of ethanol 55% (6-A1), the concentration of APIs was comparable to the case in which the tablets’ protective film was removed before two-step dissolution in the same co-solvents/water mixture composition. It is noteworthy that removing the protective film of the tablets leads to pharmaceutical formulations equivalent to the commercial syrup in terms of drug content (entry 4-A4, table 3), although manipulative procedures for film removal required manual skills.
The simplicity of preparing and reconstituting the powder resulting from crushing the two tablets is better when the composition of the first dispersing phase (5 mL) is ethanol/glycerol 40:60, corresponding to the final formulation containing 11% glycerol (as in 3-A2). In fact, the volume and viscosity of the first dispersing phase may become critical factors in refining the experimental procedures mainly when the reconstitution is performed in the pharmacy or the hospital ward, where the simplest compounding method should be chosen. In fact, a reduced volume of the first dispersing phase could make powder reconstitution more difficult as in case 6-A1 (3.6 mL, corresponding to the final concentration of 6% glycerol) in comparison to 3-A2 (5 mL). By contrast, a major content of glycerol (15%) corresponds to a remarkable increase of viscosity which does not facilitate wetting of the powder and hence time needed for dissolution.
In all cases when the powder is previously wetted with the ethanol/glycerol solution, the formulations are stable over time (48 hours/4°C) (figure 3).
 Figure 3
Figure 3 1H NMR (600 MHz, CD3OD, 25°C) spectral regions corresponding to the selected aromatic proton of ritonavir in corresponding galenic formulations 30 min (t0) (·) and 48 hours at 4°C (▲) after preparation. NMR, nuclear magnetic resonance.
ConclusionsAn efficient procedure for extemporaneous preparation, immediately before administration, of the active ingredients lopinavir/ritonavir liquid formulations from tablets has been designed. Preparative protocol combines remarkable practicality and simplicity, even in the reconstitution phase in the hospital ward, to a high level (about 90%) of content and stability of active ingredients over time (48 hours at 4°C).
Procedural optimisation phases have been efficiently assisted by a non-invasive NMR analytical methodology, the reliability of which in terms of quantitative response has been recognised for a long time.14–17
Poor solubility of the active ingredients in only water has been confirmed. The dispersion in water of crushed tablets alone has also led to lower content of active ingredients.19–21
Therefore, the two-step setup method with final 7.1% ethanol and 11% glycerol concentration is the best operating condition, providing an amount of completely dissolved APIs comparable to the commercial formulation and with high stability maintenance, at refrigerated temperature, within 48 hours. The operating instruction of lopinavir 400 mg/ritonavir 100 mg powder for oral suspension has been submitted and published on the EAHP COVID-19 Resource Centre.22
However, preparing the galenic formula in the pharmacy lab may allow an operating condition to further improve the solubilisation of active ingredients: the A1 method, characterised by a reduced volume of dispersing phase 1 with a higher percentage of ethyl alcohol, or the A4 method involving the removal of the coating film from the tablets. The latter two operating conditions led to extemporaneous galenic preparation with a usual shelf life in accordance with oral liquid formulation stability without preservative additives.
The two-step setup method ready to use in the ward may be the best choice for the hospital’s organisational needs. In fact, crushed tablets, phase 1 dispersants and water can be separately stored at room temperature before use.
What this paper addsWhat is already known on this subjectThe lopinavir/ritonavir combination has been used in the treatment of COVID-19 disease, both in the early stage and in advanced intensive care, and liquid formulations were needed for administration to passive patients (sedated and intubated).
The galenic lopinavir/ritonavir liquid formulation from crushed tablets in water, unlike the marketed formulations, seems poorly soluble due to the in-water insolubility of active ingredients.
What this study addsAn analytical nuclear magnetic resonance method for the study of galenic formula lopinavir/ritonavir liquid formulations is described.
An efficient and simple procedure is presented for extemporaneous preparation of galenic lopinavir/ritonavir liquid formulation, with 90% content and stability of active pharmaceutical ingredients over a 48 hour period stored at 4°C.
Data availability statementNo data are available.
Ethics statementsPatient consent for publicationAcknowledgmentsThe authors would like to thank the staff of the pharmacy unit of Pisa University Hospital for their cooperation and support during this very extraordinary emergency for the Italian healthcare system.
留言 (0)