
記住我
Primary immune thrombocytopenia (ITP) is an acquired immunological disorder defined as thrombocytopenia less than 100 × 109/l with no underlying cause [1]. In the general medical practice, ITP is the most common cause of severe isolated thrombocytopenia (80%). The incidence of ITP ranges from 2 to 4 cases per 100 000 person-years, with a peak incidence between 20 and 30 years of age, with a slight female predominance and a larger peak above 60 years of age with an equal sex distribution [2]. The term idiopathic thrombocytopenic purpura was changed to immune thrombocytopenia and later to primary immune thrombocytopenia in recognition of the pathogenesis and to exclude other causes of thrombocytopenia [3,4].
Devoid of a definitive confirmatory test, it is difficult to reliably establish the diagnosis without excluding congenital and acquired causes. Congenital thrombocytopenia may masquerade as ITP. Hence, without historical platelet counts from the patient or family members, ITP may be difficult to diagnose. Primary immune thrombocytopenia may also be the initial manifestation of acquired autoimmune disorders including systemic lupus erythematosus (SLE), wherein 20–30% of patients develop ITP during the course of the disease [5]. The clinical course of ITP may be very heterogeneous. Although most of the childhood ITP remit spontaneously without complications, some children bleed and develop a chronic protracted illness. Adults with ITP may enter a deep remission with a short course of corticosteroids alone or suffer a chronic relapsing disease [6,7].
Since the original description by Harrington and James W. Hollingsworth in 1950, there has been no significant advancement made in the diagnostic criteria, despite the fact that rapid strides have been made in the management of ITP [8]. In practice, after initial suspicion of ITP and detailed clinical evaluation, appropriate tests to exclude a secondary cause is done before treatment is initiated [9]. Our aim is to study the diagnostic tests available for ITP and analyze the evidence for each of them in support of the diagnosis of ITP.
MethodsWe used Medline and Cochrane databases through August 2021 and found 3797 studies reporting data on the diagnosis of ITP. To be considered for this review, the inclusion criteria encompassed the following: the article must be on primary ITP, the diagnostic test should be unequivocal in the diagnosis and the article had to deal explicitly with the diagnosis of ITP excluding other causes confounding the diagnosis. Thus, we excluded articles pertaining to thrombocytopenia including secondary ITP, inherited disorders, medication, herbal mediated, primary marrow disorders, sequestration induced and other disorders associated with hemostasis. We then selected diagnostic tests and analyzed their individual merits in support of the diagnosis of ITP.
Data extractionRandomized controlled trials, case series and abstracts published in peer reviewed journals presented in national and international meetings were reviewed. Data was extracted on author names, location, specific intervention, comparison details, outcomes and participants according to the Cochrane Handbook for Systematic Reviews of Interventions. Two authors (N.V. and R.K.) in duplicate reviewed the titles, abstracts, and full texts. Standardized data extraction form was used for data collection. Conflicts were resolved by consensus through consultation with other authors. Additional references were obtained from references retrieved by the manual search and from the bibliographies of the retrieved articles.
Results Peripheral blood smearWhenever ITP is suspected, the peripheral blood smear is examined to exclude other causes of thrombocytopenia including pseudothrombocytopenia. The blood smear is reviewed for abnormal red blood cells (schistocytes), white cells (blasts, toxic granulation of polymorphs, Döhle bodies) and platelets (clumping of platelets, rosette formation) prior to the diagnosis of ITP [10]. Congenital thrombocytopenia may be identified by changes in platelet morphology (giant platelets, gray platelets) [11]. The interpretation of blood smear may require expertise and easy access to microscopy. Digital microscopes allowing transmission of images to experts in remote locations and computer-assisted digital microscopes for evaluation of blood films are under development. Apart from the paucity of platelets, there is no diagnostic marker for ITP in peripheral blood smear.
Mean platelet volumeThis parameter suggests the bone marrow's response to platelet sequestration in ITP (normal range: 7.5–11.5 fl). An increased mean platelet volume is typically seen in the context of bone marrow response to increased platelet sequestration in ITP but this may also be seen in inherited thrombocytopenia. It is a machine-calculated ratio of plateletecrit and platelet count. The highest values are seen in ITP [12]. Another study found that the MPV larger than 12.4 fl can differentiate inherited macrothrombocytopenia as in MYH9-related disease and Bernard–Soulier syndrome from ITP with a sensitivity and specificity of almost 90% making MPV a useful parameter for differentiating inherited from immune thrombocytopenia [13]. In a study of 171 patients with thrombocytopenia, 4 out of 37 patients with ITP, the MPV was not elevated, though the highest values were found in ITP with a P value of less than 0.0001 [12]. The MPV in patients with ITP and healthy population was not very different (P value 0.76) but high MPV had better outcome in patients with ITP [14]. The MPV cannot be measured accurately with very low platelet count as the analyzer may be unable to derive the MPV because of the platelet histogram distribution. Therefore, MPV though statistically capable of differentiating disorders of thrombocytopenia, cannot be considered a diagnostic marker for ITP, especially when platelet count is extremely low.
Immature platelet fractionImmature platelet fraction (IPF) gives an indirect determination of platelet production. The IPF is usually elevated in ITP [15]. The immature platelets circulating in the blood contain residual RNA, giving an indirect determination of platelet production. The IPF is detected by flow cytometry. This may be a practical tool to differentiate thrombocytopenia of decreased production from increased destruction [16]. In a study using IPF, the mean IPF % value by Sysmex XE-2100 was found useful to predict ITP. In a study of 231 patients, 62 were diagnosed as ITP and 169 as non-ITP, the mean IPF % value for ITP was 16.39% compared with 7.69% for non-ITP patients [17]. In a study comparing 41 patients with ITP and 14 patients with thrombocytopenia from hematological malignancies on chemotherapy, the authors found the IPF to be a rapid and inexpensive automated marker for discerning the cause of thrombocytopenia. They concluded that IPF may also be useful as a potential prognostic marker for chronic ITP. The median IPF was 11.8% in patients with ITP, 7% in those with hematological malignancy and 3% in the control group (P < 0.001) [18]. The IPF may also be used to assess the response to treatment in ITP [19]. However, elevated IPF demonstrates rapid turnover of megakaryocytes within the bone marrow and is not specific for an underlying immune-mediated mechanism [20]. A lack of standardization of methods and definition of threshold values make the interpretation of IPF difficult [19].
The diagnostic utility of platelet-specific autoantibody assaysA systematic review of platelet GP IIb/IIIa and/or GP Ib/IX-specific glycoprotein-specific platelet autoantibody testing analyzed 1170 ITP patients, and 225 nonimmune thrombocytopenic controls. The glycoprotein-specific autoantibody assay sensitivity was low (50%), whereas the specificity was high (90%). The possible explanations for this low sensitivity and high specificity in ITP may be that a proportion of ITP patients have autoantibodies against other nonplatelet target antigens, such as thrombopoietin or its receptor c-Mpl, or the autoantibodies are undetectable in some patients (because of low titer or sequestration). In children with ITP platelet GP IIb/IIIa was positive in 72% [21]. There may be other pathological immune mechanisms present that are independent of platelet autoantibodies, such as cytotoxic T cells [22]. Assays for antibodies to specific platelet glycoproteins are not routinely recommended as platelet-associated IgG is elevated in both immune and nonimmune thrombocytopenia. This suggests that ITP is a heterogenous group of disorder caused by multiple mechanisms including, but not limited to, antiplatelet autoantibodies [23].
Other serological testsTesting for antinuclear antibody (ANA) or other serological tests are not recommended in children and adults with suspected ITP. The antiphospholipid antibodies (APLA), including anticardiolipin antibodies are found in about 40% of patients presenting with ITP [24,25]. The presence of APLA does not appear to affect the response to treatment in ITP [26]. A positive ANA test may be a predictor of chronicity of ITP [27]. Antithyroid antibody was seen in 8–14% of ITP patients who developed clinical hyperthyroidism [28]. Overall, direct antiglobulin testing or testing for ANA, antiphospholipid antibodies, antithyroid antibodies, or thyroid function tests may exclude other causes of thrombocytopenia when clinical suspicion exists [29,30].
Refractoriness of platelet transfusion in the diagnosisPatients with ITP are considered to have relative refractoriness to platelet transfusion because of platelet-specific immunoglobulins. Studies have tried to quantify the refractoriness by the 10 min postinfusion-corrected count increment (CCI) and the percentage platelet response (PPR), but this topic remains debatable [31]. The time to reach total body equilibrium for transfused platelets varies considerably between normal volunteers and thrombocytopenic patients [32]. A CCI of less than 7500 or a PPR of less than 30% have been used to define refractoriness. However, the evidence to support this concept is highly variable [33]. Confounding nonimmune factors associated with refractoriness frequently include fever, infection and medications [33]. Hence, platelet refractoriness cannot be used for the diagnosis of ITP.
Bone marrow examinationIn a study of 296 cases of childhood ITP, none had a change of the diagnosis after the bone marrow examination [34]. In adults, bone marrow examination did not change the diagnosis of ITP when there was isolated thrombocytopenia [35,36]. In a prospective study of 353 cases, bone marrow examination in ITP revealed normal hematopoietic elements. The authors in this study concluded that routine bone marrow examination may not be required in the diagnostic work-up of ITP [37]. Bone marrow examination is not recommended for routine evaluation in patients with typical ITP at presentation [29,38,39]. However, bone marrow examination must be considered in patients older than 60 years, those with atypical or specific clinical or hematological features, those with a poor response to standard therapy with steroid, or patients in whom splenectomy is contemplated [36,40].
Genomics in the diagnosisSequencing of relevant pathological genes known to cause thrombocytopenia may be an important diagnostic approach in ITP. As genetic studies for inherited causes of thrombocytopenia becomes more readily available, rates of detection and diagnosis of nonimmune platelet disorders will increase and be further characterized [41]. A thrombogenomics platform correctly identified the pathogenic variants in all 159 samples with a known inherited platelet dysfunction demonstrating an impressive sensitivity of 100% [42]. Whole exome and genome sequencing has established a number of genetic causes for thrombocytopenia [43]. This may help to differentiate congenital causes of thrombocytopenia from ITP. In patients with refractory ITP with atypical features, alternative causes of thrombocytopenia should be considered. Several well characterized platelet genes encode the most abundant platelet-specific proteins including glycoprotein Ibα, glycoprotein IIb and platelet factor 4. Abnormalities of the genes may help to delineate the platelet function and immunological status of the platelets [44].
The role of thrombopoietinPatients with ITP have normal or only slightly elevated thrombopoietin levels. In contrast, patients with other causes of thrombocytopenia, in particular, aplastic anemia, have markedly elevated levels of thrombopoietin (TPO) [45]. A prospective screening study of endogenous thrombopoietin levels in 205 ITP subjects showed no significant difference between circulating concentrations of TPO level in control versus patients with ITP [45]. Routine testing for TPO levels is not recommended for diagnostic purposes [46]. The role of serum TPO in predicting response to thrombopoietin receptor agonist (TPO-RA) therapy is under research [1,29].
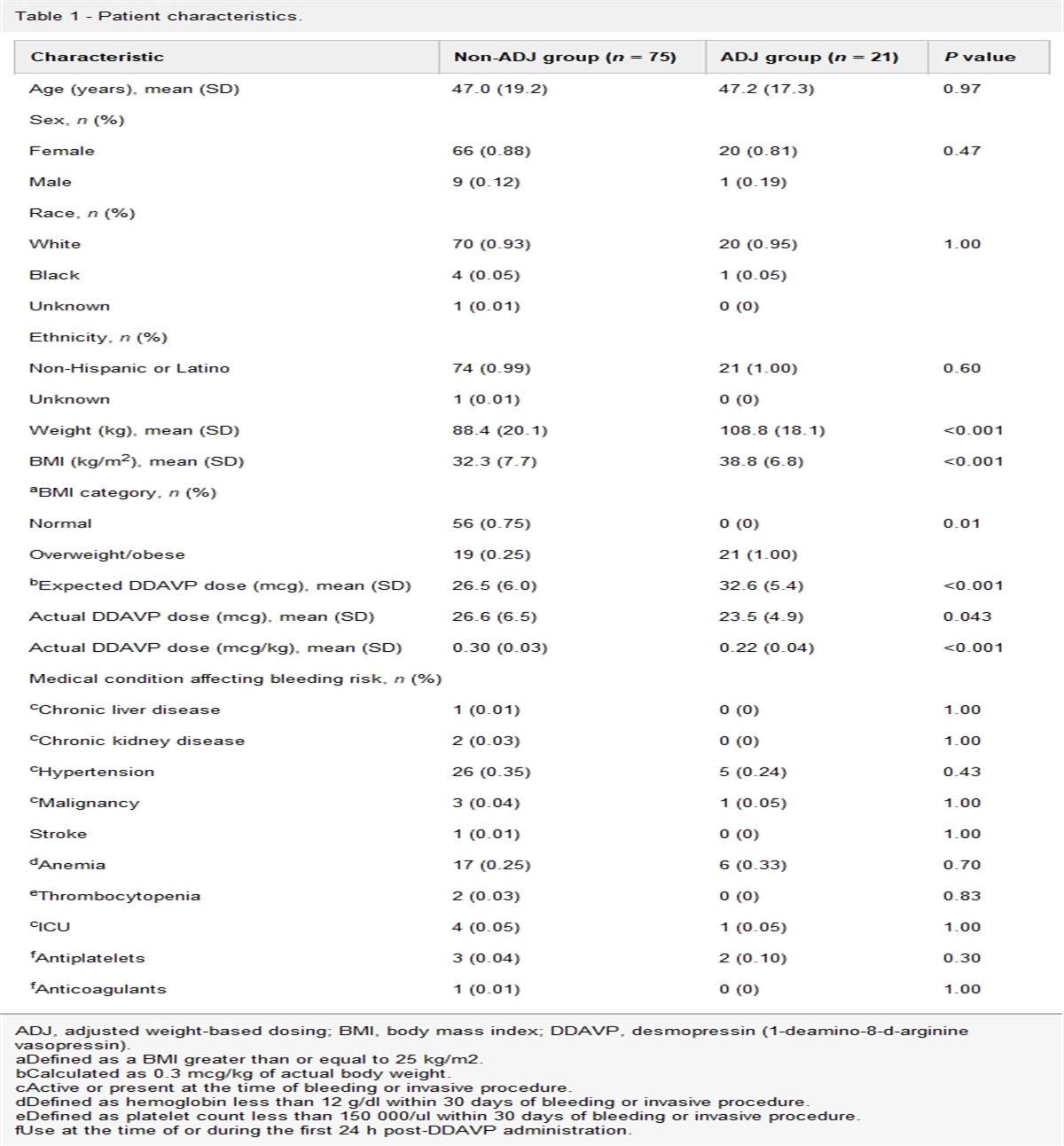
Diagnostic difficulties in immune thrombocytopeniaEstablishing the diagnosis of ITP in most patients seems relatively straightforward for experienced physicians but standardization of the diagnosis of ITP remains challenging. In our search, we found no prospective or retrospective study unequivocally confirming the diagnosis of ITP (Table 1). The need to identify secondary causes is crucial for definitive therapy and lack of agreement in the diagnostic criteria can be challenging [47]. When a panel of three hematologists provided their opinion on thrombocytopenia by reviewing case reports, there was discrepancy in the diagnosis of ITP among panelists [48].
Table 1 - Studies analyzing different parameters in immune thrombocytopenia Author Study type Patients Age group Parameter Confirms ITP Comment Akkuş et al. [14] R/P 70 Adults MPV No Good prognosis if MPV is high Jeon et al. [20] R/P 568 Adults IPF No Elevated in ITP. Cannot be used as a sole criterion Schmidt et al. [21] P/P 179 Children GPIIb/IIIa 72%+ve Prognosis is good if GPIIb/IIIa is positive Porcelijn et al. [46] P/P 72 Children/adults TPO No Not useful in diagnosis of ITP Abdurrahman et al. [38] R/P 122 Children Bone marrow No Not useful in diagnosis of ITP Gunduz et al. [39] R/P 98 Adults Bone marrow No Not useful in diagnosis of ITPITP, immune thrombocytopenia; MPV, mean platelet volume; P/P, prospective; R/P, retrospective; TPO, thrombopoietin.
One study showed improved agreement in the diagnosis of ITP when the following criteria were present: a low platelet nadir (<20 × 109/l) and a platelet count increase following treatment with intravenous immunoglobulin (IVIG) or corticosteroids but this will not exclude secondary ITP [48]. A case series found 15% of ITP patients fulfilling the diagnosis of SLE on detailed evaluation [49]. Up to 10% of patients with ITP test positive for antiphospholipid antibodies and/or lupus anticoagulant [50]. About 5–10% of patients with lymphoproliferative disorder and 10% with common variable immunodeficiency develop ITP [51]. In a large, prospective, multicenter, international registry over 15 years, 3974 children and adolescents with an initial diagnosis of primary ITP were analyzed, revisions to the diagnosis were made in 241 children within 24 months of follow-up. Ultimately, 113 patients had an unequivocal diagnosis of secondary ITP [52]. Another study found that 10 (13%) of 75 ITP patients had positive serologic findings for Epstein–Barr virus, cytomegalovirus, or rubella virus-associated immune thrombocytopenia in childhood [53]. Contrary to the expectation, initial platelet counts and the number of patients with profound thrombocytopenia (<20 × 109/l) were not significantly different between secondary and primary ITP [54,55].
Caution in accepting immune thrombocytopenia as a diagnosis of exclusionThe diagnosis of ITP needs great caution and follow-up as this may convert to other immune-mediated disorders over time [56]. One study estimated that the diagnosis of ITP was inaccurate among one in seven patients suspected of having ITP [57]. Response to ITP-specific therapy including intravenous immunoglobulin (IVIg), intravenous anti-D, steroid therapy, rituximab and splenectomy appears to be the single most diagnostic criterion [1]. However, response to these agents may occur in other conditions. In a study of 492 patients who had the diagnosis of ITP, 17% were found to have alternative diagnoses on chart review, with coding classification errors in 3%, or an alternative explanation for their thrombocytopenia consisting of 31 different diagnoses in 14%. The most common diagnoses were familial thrombocytopenia (10%), systemic lupus erythematosus (9%), hypersplenism (9%), neonatal alloimmune thrombocytopenia (7%), Wiskott–Aldrich syndrome (7%) or systemic infection (6%). In total, 16 patients (23% of the alternative diagnosis and 3% of the total population) were ultimately diagnosed with inherited platelet function disorders [58].
There are algorithms published in the medical literature to help with the diagnosis of ITP. These algorithms are aimed to describe, which test/parameter should be undertaken in case of chronic ITP but do not appear to be informative [30,59]. Some investigators have suggested that a progressive algorithm with further investigations to be performed in patients with persistent and chronic ITP. However, these algorithms are driven by the specific investigations performed and not by the possible differential diagnosis [60].
The heterogeneity of ITP is demonstrated by the bleeding symptoms. Most patients with chronic ITP with low platelet count do not bleed [4]. Some with higher platelet counts bleed spontaneously and dangerously. There is no direct correlation with the platelet count and the severity of illness in ITP. When measured systematically using an ITP-specific bleeding tool, a prospective study suggested that the frequency of grade 2 bleeding is higher than previously reported [57]. A systematic review found that 56% of patients with primary ITP experienced severe bleeding at some point during their disease course. This estimate is higher than what has been previously reported in the medical literature. Severe bleeding occurred in approximately 10% for adults out of which intracranial hemorrhage accounted for 1–1.5% of the cases [61]. In a retrospective study with 13 064 patient-years of follow-up, 3768 patients (57%) experienced at least one bleeding-related events per patient-year. The majority (58%) of bleeding-related events were treated with rescue therapy. Common bleeding categories included gastrointestinal hemorrhage, hematuria, ecchymosis and epistaxis [62]. History of mucocutaneous bleeding cannot be used to differentiate ITP from other causes of thrombocytopenia.
LimitationsThis review has certain limitations, including the small sample size of the included studies. There was lack of standardization of methods and consistency of the tests. This hinders meaningful assessment of test results. Due to the nature of the search methodology used, some literature may have been inadvertently omitted from the conferences outside of Europe and America as well as unpublished articles. The number of studies were limited to reporting and publication bias. The study is also limited by challenges of an on-line literature search.
ConclusionThe diagnosis of ITP continues to be presumptive and empirical. As there is no diagnostic test for ITP, the clinician is best served by a comprehensive physical examination, examination of the peripheral blood smear (for morphological abnormalities) and assess initial treatment response. When patients fail initial therapy, bone marrow examination, auto-immune serology (to rule-out an underlying connective tissue disorder), iatrogenic and infectious disease evaluations should be considered, unless an obvious cause for the thrombocytopenia becomes evident. Identifying the underlying cause of thrombocytopenia is crucial for the appropriate management. There appears to be no agreement regarding the tests needed to complete the diagnostic work-up. Future research in diagnostic testing for ITP is needed to prevent morbidity. Identifying the underlying cause of thrombocytopenia is crucial for the management of ITP
AcknowledgmentsFinancial disclosure: no specific funding was received from any public, commercial, or not-for-profit sectors to carry out the work described in this article.
Informed consent: there was no informed consent required for this article.
Author contributions: N.V., I.R., M.K., and L.S. discussed the project and the main conceptual ideas and developed the framework. N.V. and R.K. collected the data. D.L. supervised the project. N.V. wrote the initial manuscript. A.M. reviewed the final version before submission. All authors discussed the results and contributed to the final manuscript.
Data availability: the authors declare that data supporting the findings of this study are available within the article.
Conflict of InterestThere are no conflicts of interest.
References 1. Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186. 2. Marieke Schoonen W, Kucera G, Coalson J, Li L, Rutstein M, Mowat F, et al. Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database. Br J Haematol 2009; 145:235–244. 3. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393. 4. Ruggeri M, Fortuna S, Rodeghiero F. Heterogeneity of terminology and clinical definitions in adult idiopathic thrombocytopenic purpura: a critical appraisal from a systematic review of the literature. Haematologica 2008; 93:98–103. 5. Boumpas DT, Austin HA, Fessler BJ, Balow JE, Klippel JH, Lockshin MD. Systemic lupus erythematosus: emerging concepts: part 1: renal, neuropsychiatric, cardiovascular, pulmonary, and hematologic disease. Ann Intern Med 1995; 122:940–950. 6. Wiley Online Library, Arnold DM. Platelet count or bleeding as the outcome in ITP trials? 2012. 7. Neunert CE, Buchanan GR, Imbach P, Bolton-Maggs PH, Bennett CM, Neufeld EJ, et al. Severe hemorrhage in children with newly diagnosed immune thrombocytopenic purpura. Blood 2008; 112:4003–4008. 8. Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10. 9. George JN, Woolf SH, Raskob GE, Wasser JS, Aledort LM, Ballem PJ, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 1996; 88:3–40. 10. Adewoyin A. Peripheral blood film-a review. Ann Ibadan Postgrad Med 2014; 12:71–79. 11. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004; 103:390–398. 12. Kickler TS, Oguni S, Borowitz MJ. A clinical evaluation of high fluorescent platelet fraction percentage in thrombocytopenia. Am J Clin Pathol 2006; 125:282–287. 13. Noris P, Klersy C, Zecca M, Arcaini L, Pecci A, Melazzini F, et al. Platelet size distinguishes between inherited macrothrombocytopenias and immune thrombocytopenia. J Thromb Haemost 2009; 7:2131–2136. 14. Akkuş E, Fidan Ç, Demirci G, Kuştaş AA, Yüksel M. Mean platelet volume and response to the first line therapy in newly diagnosed adult immune thrombocytopenia patients: a retrospective study. Turk J Med Sci 2020; 50:798–803. 15. Jiménez MM, Guedán MA, Martín LM, Campos JS, Martínez IR, Vilella CT. Measurement of reticulated platelets by simple flow cytometry: an indirect thrombocytopoietic marker. Eur J Intern Med 2006; 17:541–544. 16. Dusse LMS, Freitas LG. Clinical applicability of reticulated platelets. Clin Chim Acta 2015; 439:143–147. 17. Naz A, Mukry SN, Shaikh MR, Bukhari AR, Shamsi TS. Importance of immature platelet fraction as predictor of immune thrombocytopenic purpura. Pak J Med Sci 2016; 32:575. 18. Adly AAM, Ragab IA, Ismail EAR, Farahat MM. Evaluation of the immature platelet fraction in the diagnosis and prognosis of childhood immune thrombocytopenia. Platelets 2015; 26:645–650. 19. Thomas-Kaskel AK, Mattern D, Köhler G, Finke J, Behringer D. Reticulated platelet counts correlate with treatment response in patients with idiopathic thrombocytopenic purpura and help identify the complex causes of thrombocytopenia in patients after allogeneic hematopoietic stem cell transplantation. Cytometry B Clin Cytom 2007; 72:241–248. 20. Jeon MJ, Yu ES, Kang K-W, Lee B-H, Park Y, Lee SR, et al. Immature platelet fraction based diagnostic predictive scoring model for immune thrombocytopenia. Korean J Intern Med 2020; 35:970. 21. Schmidt DE, Heitink-Polle KM, Porcelijn L, van der Schoot CE, Vidarsson G, Bruin MC, de Haas M. Antiplatelet antibodies in childhood immune thrombocytopenia: prevalence and prognostic implications. J Thromb Haemost 2020; 18:1210–1220. 22. Nazy I, Kelton JG, Moore JC, Clare R, Horsewood P, Smith JW, et al. Autoantibodies to thrombopoietin and the thrombopoietin receptor in patients with immune thrombocytopenia. Br JHaematol 2018; 181:234–241. 23. Olsson B, Andersson P-O, Jernås M, Jacobsson S, Carlsson B, Carlsson LM, Wadenvik H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med 2003; 9:1123–1124. 24. Brighton T, Evans S, Castaldi P, Chesterman C, Chong B. Prospective evaluation of the clinical usefulness of an antigen. Blood 1996; 88:194–201. 25. McMillan R, Wang L, Tani P. Prospective evaluation of the immunobead assay for the diagnosis of adult chronic immune thrombocytopenic purpura (ITP). J Thromb Haemost 2003; 1:485–491. 26. Pierrot-Deseilligny C, Khellaf M, Gouault M, Intrator L, Michel M, Bierling P, Godeau B. Prevalence and clinical significance of elevated antiphospholipid antibodies in adults with immune thrombocytopenic purpura. Am Soc Hematol 2006; 84:4203–4208. 27. Altintas A, Ozel A, Okur N, Okur N, Cil T, Pasa S, Ayyildiz O. Prevalence and clinical significance of elevated antinuclear antibody test in children and adult patients with idiopathic thrombocytopenic purpura. J Thromb Thromb 2007; 24:163–168. 28. Liebman H. Other immune thrombocytopenias. In Seminars in hematology. Elsevier; 2007: pp. S24–S34. 29. Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207. 30. Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 2019; 3:3780–3817. 31. O’connell B, Lee E, Schiffer C. The value of 10-min posttransfusion platelet counts. Transfusion 1988; 28:66–67. 32. Brubaker DB, Marcus C, Holmes E. Intravascular and total body platelet equilibrium in healthy volunteers and in thrombocytopenic patients transfused with single donor platelets. Am J Hematol 1998; 58:165–176. 33. Bishop J, Matthews J, Yuen K, McGrath K, Wolf M, Szer J. The definition of refractoriness to platelet transfusions. Transfus Med 1992; 2:35–41. 34. Watts RG. Idiopathic thrombocytopenic purpura: a 10-year natural history study at the childrens hospital of alabama. Clin Pediatr 2004; 43:691–702. 35. Westerman DA, Grigg AP. The diagnosis of idiopathic thrombocytopenic purpura in adults: does bone marrow biopsy have a place? Med J Australia 1999; 170:216–217. 36. Mak Y, Yu P, Chan C, Chu Y. The management of isolated thrombocytopenia in Chinese adults: does bone marrow examination have a role at presentation? Clin Lab Haematol 2000; 22:355–358. 37. Purohit A, Aggarwal M, Singh PK, Mahapatra M, Seth T, Tyagi S, et al. Re-evaluation of need for bone marrow examination in patients with isolated thrombocytopenia contributors. Ind J Hematol Blood Transfus 2016; 32:193–196. 38. Abdurrahman K, Hasan K, Muhsen A. Is bone marrow examination justified in isolated childhood thrombocytopenia? Qatar Med J 2012; 2012:14. 39. Gunduz E, Kivanc BK, Arik D, İsiksoy S, Bal C, Akay OM. Bone marrow examination in patients with immune thrombocytopenia: is there anything different in older patients? Eur J Haematol 2014; 93:157–160. 40. Cooper N, Ghanima W. Immune thrombocytopenia. New Engl J Med 2019; 381:945–955. 41. Fiore M, Pillois X, Lorrain S, Bernard M-A, Moore N, Sié P, et al. A diagnostic approach that may help to discriminate inherited thrombocytopenia from chronic immune thrombocytopenia in adult patients. Platelets 2016; 27:555–562. 42. Lentaigne C, Freson K, Laffan MA, Turro E, Ouwehand WH. Inherited platelet disorders: toward DNA-based diagnosis. Blood 2016; 127:2814–2823. 43. Macaulay IC, Carr P, Gusnanto A, Ouwehand WH, Fitzgerald D, Watkins NA. Platelet genomics and proteomics in human health and disease. J Clin Invest 2005; 115:3370–3377. 44. Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL, Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood 2003; 101:2285–2293. 45. Aledort LM, Hayward CP, Chen MG, Nichol JL, Bussel J. Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am J Hematol 2004; 76:205–213. 46. Porcelijn L, Folman CC, Bossers B, Huiskes E, Overbeeke MA, vd Schoot CE, et al. The diagnostic value of thrombopoietin level measurements in thrombocytopenia. Thromb Haemost 1998; 79:1101–1105. 47. Kojouri K, Perdue J, Medina P, George J. Occult quinine-induced thrombocytopenia. J Oklahoma State Med Assoc 2000; 93:519–521. 48. Salib M, Clayden R, Clare R, Wang G, Warkentin TE, Crowther MA, et al. Difficulties in establishing the diagnosis of immune thrombocytopenia: an agreement study. Am J Hematol 2016; 91:E327–E329. 49. Kumar S, Nair S, Rajam L. Case series of pediatric systemic lupus erythematosus from Kerala: comparison with other Indian series. Int J Rheum Dis 2010; 13:391–395. 50. Stasi R, Stipa E, Masi M, Oliva F, Sciarra A, Perrotti A, et al. Prevalence and clinical significance of elevated antiphospholipid antibodies in patients with idiopathic thrombocytopenic purpura. Blood 1994; 84:4203–4208. 51. Grainger JD, Bolton-Maggs PH, Godeau B, Bussel J, Donato H, Elalfy M, et al. Diagnosis and management of chronic ITP: comments from an ICIS expert group. Ann Hematol 2010; 89:11–17. 52. Schifferli A, Heiri A, Imbach P, Holzhauer S, Seidel MG, Nugent D, et al. Misdiagnosed thrombocytopenia in children and adolescents: analysis of the Pediatric and Adult Registry on Chronic ITP. Blood Adv 2021; 5:1617–1626. 53. Yenicesu İ, Yetgin S, Özyürek E, Aslan D. Virus-associated immune thrombocytopenic purpura in childhood. Pediatr Hematol Oncol 2002; 19:433–437. 54. Ayesh MH, Alawneh K, Khassawneh B, Khader Y, Kasasbeh A. Adult primary and secondary immune thrombocytopenic purpura: a comparative analysis of characteristics and clinical course. Clin Appl Thromb/Hemost 2013; 19:327–330. 55. Rotz SJ, Ware RE, Kumar A. Diagnosis and management of chronic and refractory immune cytopenias in children, adolescents, and young adults. Pediatr Blood Cancer 2018; 65:e27260. 56. Tamaddoni A, Yousefghahari B, Khani A, Esmaeilidooki M, Sawadkouhi RB, Mohammadzadeh I. Isolated thrombocytopenia; a report of a rare presentation of childhood systemic lupus erythematosus (SLE). Caspian J Intern Med 2015; 6:174. 57. Arnold DM, Nazy I, Clare R, Jaffer AM, Aubie B, Li N, Kelton JG. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP Registry. Blood Adv 2017; 1:2414–2420. 58. Bryant N, Watts R. Thrombocytopenic syndromes masquerading as childhood immune thrombocytopenic purpura. Clin Pediatr 2011; 50:225–230. 59. De Mattia D, Del Vecchio GC, Russo G, De Santis A, Ramenghi U, Notarangelo L, et al. Management of chronic childhood immune thrombocytopenic purpura: AIEOP consensus guidelines. Acta Haematol 2010; 123:96–109. 60. Consolini R, Costagliola G, Spatafora D. The centenary of immune thrombocytopenia—part 2: revising diagnostic and therapeutic approach. Front Pediatr 2017; 5:179. 61. Neunert C, Noroozi N, Norman G, Buchanan G, Goy J, Nazi I, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost 2015; 13:457–464. 62. Altomare I, Cetin K, Wetten S, Wasser JS. Rate of bleeding-related episodes in adult patients with primary immune thrombocytopenia: a retrospective cohort study using a large administrative medical claims database in the US. Clin Epidemiol 2016; 8:231.
留言 (0)