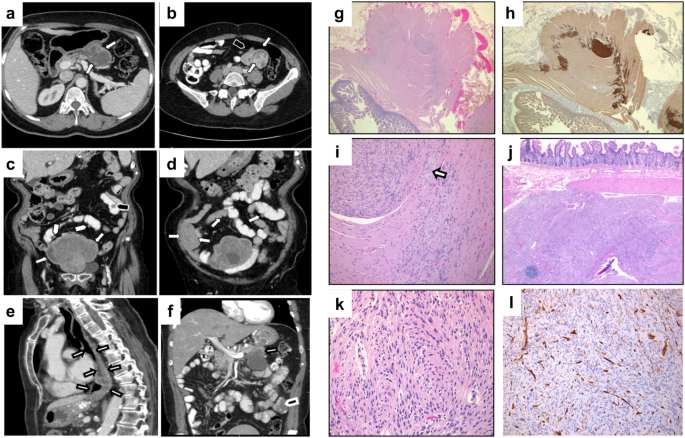
Human subjects and clinical assessments
This research was supported by the Clinical Research Center, Scientific Research and Planning Department at Ninth People’s Hospital (SH9H), Shanghai Jiao Tong University School of Medicine (JYHJB01). The human samples used in this research project were obtained with evaluation and approval from the Institutional Ethics Committee of Ninth People’s Hospital (SH9H-2020-T99-3), and all ethical guidelines conformed to the 2008 Helsinki Declaration. The project was registered under the Ninth People’s Hospital Tissue Bank (C01). All patients (or the parents of patients) and family members provided written informed consent, and they also consented to have their photographs and clinical data used for publication. Five affected individuals and eight unaffected family members from the pedigree were included in this study. When participants were enrolled in the rare diseases research project, ancillary study procedures to measure aBMD, X-ray, and CT scans of the maxillofacial and lower extremities were performed. aBMD was measured by DXA using a Lunar iDXA X-ray bone densitometer system (GE Healthcare, Chicago, IL, USA). The aBMD, X-ray and CT scans were performed by qualified technologists and interpreted by radiologists who were not involved in the design or implementation of the study. Bone turnover markers in plasma were measured by the certified clinical diagnostic laboratory of Ninth People’s Hospital. There are no reference ranges for bone turnover markers in children from Ninth People’s Hospital, so we used the reference intervals recommended by the journal Bone52.
Mice
This project has been assessed favorably by the Institutional Animal Care and Use Committee from Ninth People’s Hospital (JY-IACUC): the IACUC deemed that the abovementioned project complies with standard ethical regulations (SH9H-2020-A42-1). All animal work was approved and conducted according to ARRIVE guidelines. C57BL/6 mice were purchased from JieSiJie Laboratory Animal Corporation (Shanghai, China). Ano5 KO mice (C57BL/6-Ano5<tm1Itak>) were obtained from RIKEN BioResource Center (Kyoto, Japan) and maintained at the Animal Resources Center at Ninth People’s Hospital under specific pathogen-free (SPF) conditions; they were backcrossed with C57BL/6J mice. The genomic DNA from KO mice was amplified by PCR using a forward primer (5′-GGTTGTATTGGTTCTTAAATTGTGG) and two reverse primers (5′-AACCGAAGACTGTCACATGTGGAAT for the 257 bp-WT allele and 5′-AATTCATTCTCGATTCTTGATGG for the 407 bp-mutant allele) as described. Both male and female mice were used in this study; age- and sex-matched 4- to 6-week-old mice were used for in vivo studies, and 6- to 8-week-old mice were used for in vitro studies. For PTH-treated Ano5−/− mice, recombinant human PTH 1–34 (Beyotime, Shanghai, China) (80 µg/kg) and vehicle (NaCl) subcutaneous injections were administered 5 days per week over 3 weeks when the mice were 5–8 weeks old. Mice were maintained with a 12 h light/dark cycle and were fed standard chow. Cohoused animals were used for in vivo analyses. Littermates were used where indicated.
Cell culture
Human PBMCs were isolated from the peripheral blood of patients and controls using Ficoll-Paque PLUS (Cytiva, Marlborough, MA, USA) followed by standard operating procedures. PBMCs were cultured in Roswell Park Memorial Institute (RPMI) 1640 media (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Gaithersburg, MD, USA) and 1% penicillin and streptomycin (Gibco, Gaithersburg, MD, USA). Calvarial osteoblasts were prepared and cultured from postnatal day 1 mouse calvariae. The calvariae were digested in PBS (HyClone, Logan, UT, USA) containing 0.02% type II collagenase (Sigma Aldrich, St. Louis, MO, USA) and 0.05% trypsin for 10 min at 37 °C with shaking. BMSCs and BMMs were obtained from cultures of bone marrow collected from 4- to 6-week-old female mouse tibias and femurs as described53. Calvarial osteoblasts, BMSCs and BMMs were cultured in α-MEM (Gibco, Gaithersburg, MD, USA) supplemented with 10% FBS (Gibco, Gaithersburg, MD, USA) and 1% penicillin/streptomycin (1% p/s) (Gibco, Gaithersburg, MD, USA). All cells were cultured at 37 °C in a humidified 5% CO2 incubator. FBS was heat-inactivated before use.
WES
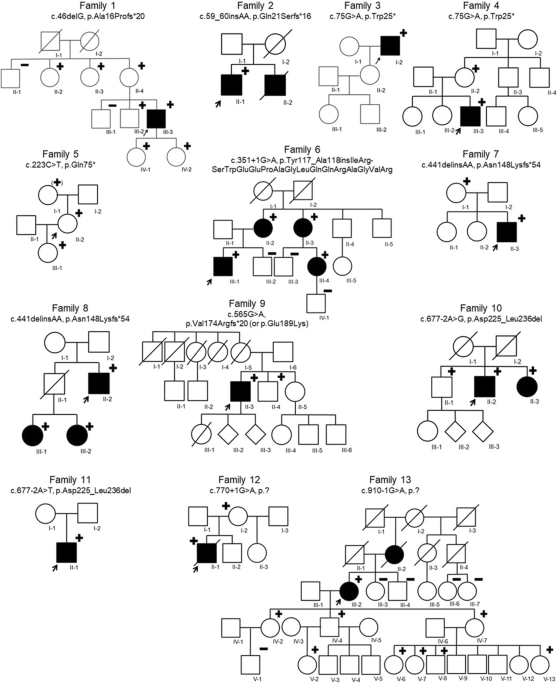
A Blood Genomic DNA Mini Kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from participant peripheral blood samples. The concentration and purity of the gDNA were determined by a spectrophotometer (NanoDrop, Thermo Fisher Scientific, Waltham, MA, USA). WES was further used to detect potential causal single nucleotide variants, insertions, and deletions (indels) in 4 patients (II4, III6, III10, IV5) and 4 controls (II5, II10, III5, III8) in the family. The hybridization capture procedure was performed with an Agilent SureSelect Illumina NovaSeq 6000 instrument (Illumina, San Diego, CA, USA) with the 180–200 bp paired-end read module. The mean coverage depth was 100×, with ~99.5% coverage of the target region. Clean reads were aligned to the human reference genome (hg19) using the Burrows–Wheeler Aligner (BWA; v.0.7.12). Whole-genome SNP scanning was performed using the Illumina Asian Screening Array. Linkage analysis was performed by the parametric linkage analysis package of MERLIN (ver. 1.1.2). Variants were filtered by the following criteria. Variants in the intronic, upstream, and downstream sites were removed, and those in the exonic and splicing functional regions were retained. New variants were classified according to the American College of Medical Genetics (ACMG) and Genomics criteria as being pathogenic, likely pathogenic, or of uncertain clinical significance, and variants classified as benign, or likely benign, were excluded. Variants were not considered if they had a read depth < 5× and a minor allele frequency (MAF) > 0.005 in databases including the dbSNP138, 1000 Genomes Project, Complete Genomics 69, Exome Variant Server, and Exome Sequencing Project databases. To assess the variant novelty, the Human Gene Mutation Database and ClinVar were used. GDD patients exhibit AD inheritance, so autosomal recessive (AR) inheritance pattern variants and variants carried by normal controls were also excluded. Thus, 9 new genetic variants were considered after the initial filtering step, which is listed in Supplementary Table 1.
Sanger sequencing
Primer3 software was used to design primers for the 9 pathogenic variants detected. Nine variants were subsequently validated by Sanger sequencing of 2 controls (II2, II7). According to the GDD AD inheritance pattern, variants carried by normal controls were also excluded, and ANO5, NANOS1, and WDR90 gene variants were determined. Again, we performed Sanger sequencing to confirm the 3 variants in 1 patient (IV6) and 2 other controls (III14, III15). After filtering the criteria, the new ANO5 variant was ultimately considered a disease-causing mutation, and the results and detailed primers of Sanger sequencing are listed in Supplementary Tables 2 and 4.
µCT
µCT of mandibles and femurs was performed using a micro-CT (Skyscan1176, USA, Bruker Siemens Inveon, Eschborn, Germany) with the following parameter settings: source voltage, 50 kV; source current, 450–500 μA; AI, 0.5 mm filter; pixel size, 9 μm; rotation step, 0.4°. Mandibles and femurs from 4- and 8-week-old male WT mice and Ano5−/− mice were dissected, cleaned of soft tissue, and wrapped in PBS-soaked gauze. Threshold segmentation of bone from marrow and soft tissue was performed in conjunction with a constrained Gaussian filter to reduce noise. Mandibular bone parameters were measured by analyzing ~400 slices of trabecular bone under the first molar. Trabecular bone parameters were measured by analyzing 201 slices of the proximal metaphysis. Cortical bone parameters were measured by analyzing 51 slices in the mid-diaphysis, where the central portion was between the proximal and distal ends of the femur. Analyses were performed in agreement with guidelines for the assessment of bone microstructure in rodents using µCT54.
Histology and histomorphometry
Histology and histomorphometry were performed as previously described55. Briefly, the mouse mandibles and femurs from 8-week-old mice were fixed with 10% neutral buffered formalin followed by decalcification in 10% EDTA for 3 weeks, paraffin embedding, TRAP staining and OCN immunohistochemistry. OCN was detected using a polyclonal OCN antibody (Cat# 23418-1-AP, 1:500 dilution, Proteintech, Sankt Leon-Rot, Germany) for 2 h, followed by Alexa 488-labeled secondary antibody (1:200 dilution, Cell Signaling Technology, Danvers, MA, USA) for 60 min. As indicated, DAPI staining (Sigma Aldrich, St. Louis, MO, USA) was also performed. Undecalcified femurs were embedded in paraffin for sectioning. Von Kossa staining was performed to determine osteoblast and osteoclast parameters. To determine the MAR, 25 mg/kg Alizarin red (Sigma Aldrich, St. Louis, MO, USA) was injected at 14 days, and 20 mg/kg calcein (Sigma Aldrich, St. Louis, MO, USA) was injected 7 days before bone collection. The indices were measured and analyzed using Bioquant Osteo 2009 v9.0 (Bioquant, Nashville, TN, USA). Bone histomorphometric parameters were calculated according to the standardized nomenclature for bone histomorphometry56. Pannoramic 250 Flash was used for slide scanning (3DHISTECH, Budapest, Hungary). An Olympus Fluoview 1000 laser confocal imaging system (Olympus, Shinjuku City, Tokyo, Japan) was used for microscope photography. Histological viewing and analysis were performed with Case Viewer (3DHISTECH, Budapest, Hungary) and ImageJ2/FIJI (NIH).
Cell proliferation assay
Cells were seeded at 1 × 104 cells per well in 96-well plates, and proliferation was determined using the cell proliferation reagent Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions.
In vitro osteoclast differentiation
Mature osteoclasts were generated as previously described57. Mouse BMMs or human PBMCs were cultured in α-MEM (Gibco, Gaithersburg, MD, USA) containing 10% FBS (Gibco, Gaithersburg, MD, USA) supplemented with 30 ng/mL recombinant mouse/human M-CSF and 50 ng/mL recombinant mouse/human RANKL (R&D Systems, Minneapolis, MN, USA) for 5–7 days in tissue culture dishes to induce osteoclast formation. Mature osteoclasts were characterized by staining for TRAP activity using the Acid Phosphatase Leukocyte Kit (Sigma Aldrich, St. Louis, MO, USA), and TRAP-positive multinucleated cells (>3 nuclei/cell) were counted.
In vitro osteoblast differentiation
For osteogenesis, BMSCs or calvarial osteoblasts were seeded at 2.5 × 104 cells per cm2 in 6- or 48-well tissue culture plates. The next day, the cells were transferred to osteogenic media: α-MEM medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% FBS (Gibco, Gaithersburg, MD, USA), 1% P/S (Gibco, Gaithersburg, MD, USA), 50 µg/mL l-ascorbic acid (Sigma Aldrich, St. Louis, MO, USA) and 5 mM β-glycerol phosphate (Sigma Aldrich, St. Louis, MO, USA). RNA isolation was performed at 0, 3 and 7 days. ALP staining was performed at 7 days using the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime, Shanghai, China), and Alizarin red staining (Sigma Aldrich, St. Louis, MO, USA) of minerals was conducted at 21 days according to the manufacturer’s instructions.
Lentiviral gene transduction
The Ano5 gene was subcloned into the pCDH-CMV-MCS-EF1-GFP-puro (CD513B-1) lentiviral vector by Shanghai IBS Biological Technology Co., Ltd. according to the manufacturer’s instructions. Ano5−/− BMMs and calvarial osteoblasts were transduced with recombinant lentiviral particles in the presence of 8 mg/mL polybrene (IBS Bio, Shanghai, China). Two days after lentivirus transduction, the cells were selected for approximately one week with 2–4 µg/mL puromycin (Sigma Aldrich, St. Louis, MO, USA) in the culture media. The expression of the recombinant RNA was confirmed by qPCR.
RNA extraction and gene expression analysis
RNA was isolated from cultured cells with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified using an AxyPrep Multisource RNA Miniprep Kit (Axygen, USA). RNA quality was confirmed using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Mouse complementary DNA (cDNA) was reverse-transcribed from 1 μg total RNA with PrimeScript RT Master Mix (Takara Bio, Kusatsu, Shiga, Japan). qPCR was performed in triplicate using TB Green Premix Ex Taq (Takara Bio, Kusatsu, Shiga, Japan) according to the manufacturer’s instructions. For each transcript examined, mRNA expression was normalized to Actin. Primer sequences for each gene are provided in Supplementary Table 3.
Calcium oscillations
A total of 1 × 104 BMMs or human PBMCs were seeded on 35-mm confocal dishes and cultured with 30 ng/mL M-CSF (R&D Systems, Minneapolis, MN, USA) in the presence of 50 ng/mL RANKL (R&D Systems, Minneapolis, MN, USA) or without RANKL as controls for 72 h. BMSCs (2 × 104) were cultured on confocal dishes with osteogenic media for 7 days. The assay buffer consisted of Hank’s buffered salt solution (Gibco, Gaithersburg, MD, USA, without CaCl2), 2% FCS, and 1 mM probenecid solution (Invitrogen, Carlsbad, CA, USA) (pH 7.4). The cells were then incubated in the presence of 5 µM Fluo-4 AM (Invitrogen, Carlsbad, CA, USA) and 0.05% Pluronic F-127 (Invitrogen, Carlsbad, CA, USA) in the dark at 37 °C for 45 min in assay buffer. The cells were washed twice with assay buffer and then incubated in fresh assay buffer for 20 min. The cells were viewed on the inverted stage of a Leica TCS SP8 confocal microscope (Leica Biosystems, Wetzlar, Germany). At an excitation wavelength of 488 nm, Fluo-4 was analyzed simultaneously at 5 s intervals for BMMs or human PBMCs and at 10 s intervals for BMSCs. The increase from the basal level was determined by adding 10 μM ionomycin (Abcam, Cambridge, UK). Peak analysis was performed to obtain parameters with ImageJ2/FIJI (NIH).
Flow cytometric detection of phosphatidylserine (PS)
To induce PS translocation, BMMs and primary osteoblasts were seeded at a density of 10 × 4 cells/cm2, cultured, and differentiated for 48 h and 7 days, respectively. Cells were harvested by trypsinization, washed first in growth medium and then washed in PBS (HyClone, Logan, UT, USA) supplemented with 0.5 mM CaCl2. Subsequently, the cells were treated with 10 mM ionomycin (Abcam, Cambridge, UK) or ethanol for 5 min or 2.5 μM staurosporine (Abcam, Cambridge, UK) for 8 h at 37 °C. Ionomycin was removed by washing in ice-cold PBS. Treated cells were then washed in annexin-binding buffer and stained with Alexa Fluor® 488 annexin V and propidium iodide (PI) (Invitrogen, Carlsbad, CA, USA) according to standard operating procedures. Gating strategy: (1) implement FSC/SSC gate for living cells, (2) define the gate for FITC- or FITC + cells, (3) apply this gate to all samples. Flow cytometric analysis was conducted using a BD LSR Fortessa (Becton Dickinson, Heidelberg, Germany), and the data were processed with FlowJo v.10.7.
Confocal fluorescence microscopy
Cells were seeded in cell culture dishes with glass bottoms (Greiner Bio, Monroe, NC, USA). Live cell imaging was performed using a Leica TCS SP8 confocal laser scanning microscope (Leica Biosystems, Wetzlar, Germany) equipped with ×40 oil immersion objectives. Final images represent the average of 4–12 acquisitions. No filtering was applied.
Western blot analysis
Cells were lysed in RIPA buffer in the presence of phosphatase and protease inhibitor cocktails (Thermo Fisher Scientific, Waltham, MA, USA). A standard protocol was used for Western blotting. In brief, samples were separated on 4–20% SurePage gels (GenScript, Piscataway, NJ, USA) and transferred onto a 0.45-μm PVDF membrane (Thermo Fisher Scientific, Waltham, MA, USA). Antigen detection was performed using antibodies directed against GAPDH (1:1000 dilution, Cat# 5174), β-Actin (1:1000 dilution, Cat# 3700), β-Catenin (1:1000 dilution, Cat# 8480), NFATc1 (1:1000 dilution, Cat# 8032), c-Fos (1:1000 dilution, Cat# 2250), GSK-3β (1:1000 dilution, Cat# 12456), Axin1 (1:1000 dilution, Cat# 2087), and Dvl2 (1:1000 dilution, Cat# 3224) from Cell Signaling Technology, Danvers, MA, USA; WNT1 (1:1000 dilution, Cat# abs131412) from Absin Bioscience Inc., Shanghai, China; ANO5/TMEM16E (1:100 dilution, Cat# LS‑C330335) from LSBio, Seattle, WA, USA. The bound primary antibodies were detected with anti-mouse IgG (H + L) (Cat# 5257) and anti-rabbit IgG (H + L) (DyLight™ 800 4× PEG Conjugate) (Cat# 5151) (diluted to 1:2000; Cell Signaling Technology, Danvers, MA, USA) using the Odyssey CLx system (LI-COR Biosciences, Lincoln, NE, USA). Western blot images were quantified with Image Studio Lite (LI-COR Biosciences, Lincoln, NE, USA). All blots or gels were derived from the same experiment and processed in parallel. Uncropped and unprocessed scans of blots in this study are provided in Supplementary Fig. 5.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.0 software. We used the unpaired two-tailed Student’s t test for comparisons between two groups and the multiple t test or two-way ANOVA with Tukey’s correction for multiple comparisons. Bar graphs present the mean ± standard error of the mean (SEM). No statistical methods were used to determine the sample size, and incomplete data were excluded from the analysis performed in this manuscript. Statistical details about the sample size (n) and p value are reported in the figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.



留言 (0)