
記住我

The Botswana National Comprehensive School Eye Health Program (‘Pono Yame’).
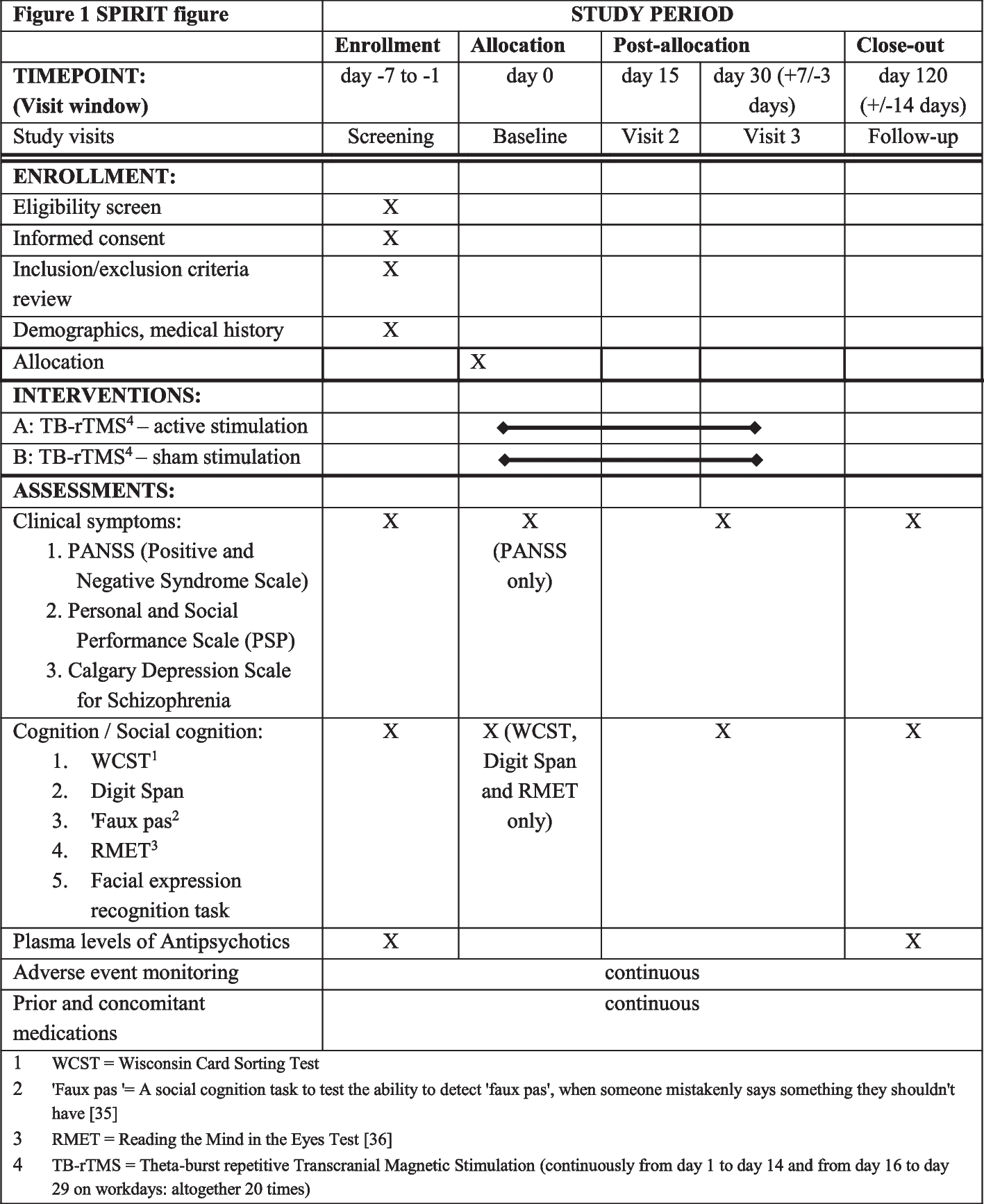
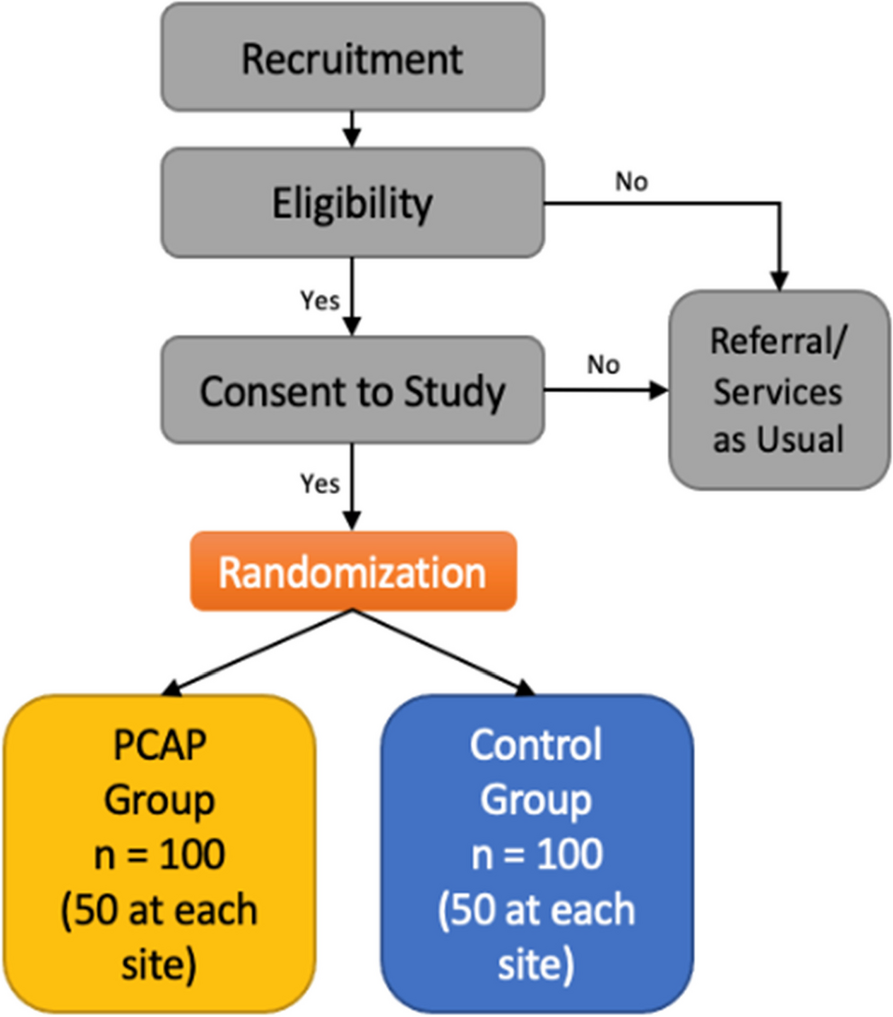
Eligibility criteriaReminder messages will be sent to the registered mobile phone numbers of parents/guardians of children who test positive at screening and are referred on to clinic in the Pono Yame MoH/Peek Vision school screening programme in 2022. Provision of a mobile number is a pre-condition of entry into the screening programme, although parents/guardians are able to supply the number of a friend or relative so in practice this stipulation does not exclude any children.
Reminders will be sent in English and Setswana; spoken by >96% of the local population. The screening programme routinely collects data on preferred language, and reminders will be sent in the preferred tongue. Those who list any language other than English of Setswana will receive the reminder in both Setswana and English. The reminder will also be sent in both languages to those where data on language is not available for any reason. We will perform a secondary analysis that excludes these participants, but they will be included in the primary analysis.
Who will take informed consent?The interventions represent minor modifications to existing routine processes and present negligible risk to participants. Obtaining consent would introduce burdens to the participant that are greater than the intervention itself. As such, we will not seek informed consent. This approach has been approved by the LSHTM and University of Botswana ethics committees, and follows the precedent set by three previous RCTs testing SMS reminder messages [12, 13, 23].
Additional consent provisions for collection and use of participant data and biological specimensAll parents/guardians are verbally informed that their children will partake in the Pono Yame vision screening programme. They are also asked to provide written opt-out consent for the use of their children’s sociodemographic data for research and sharing purposes. Care will not be compromised in any way for those participants whose parents do not provide consent.
InterventionsExplanation for the choice of comparatorsThe standard SMS message presented in Table 1 is routinely sent to the registered mobile phone of parents/guardians of children referred on for refractive services in all Peek programmes. This is the control arm.
Table 1 Control and intervention reminder messagesIntervention description Process of developing the intervention SMS and voice reminder messagesWe aimed to use an established framework to identify a theory-informed set of behaviour change principles to guide the development of our reminder messages. We elected to use Dolan and colleagues’ MINDSPACE framework [24], developed in conjunction with the Institute for Government. This framework brings together insights from behavioural economics research that can be used to develop brief healthcare interventions (Table 2). The framework has been endorsed by the Behavioural Science and Public Health Network [25], the London School of Economics Behavioral Economics Playbook for behaviour change [10] and the Health Foundation in their guidance on behavioural insights in health care [11].
Table 2 The ‘MINDSPACE’ framework and application for phone-based reminder messagesWhilst SMS and voice messages can include messenger, incentives, norms, defaults, salience, emotional appeals, commitments, and ego, they are less able to ‘prime’ recipients using subconscious cues. In addition to these principles, we also looked to the specific guidance on sending effective phone messages to reduce clinic non-attendance produced by Public Health England in 2020, based on their review of the international literature [26]. Their key messages are summarised below:
Messages should be clear, brief and well-formatted, with essential information only.
Use line breaks to make the message easier to read.
Personalise the text messages to include the recipient’s name if local systems allow.
Keep messages to 480 characters (3 standard text messages) in length.
Include the date, time, and location of the appointment, as well as any special instructions, and contact phone number (if different to the number the text message is sent from).
Write out the day of the week and the month in dates. For example, ‘Monday 23 March’.
GP endorsement can encourage people to take screening more seriously.
One researcher (LA) drafted an initial SMS that included all of the eight relevant MINDSPACE behavioural economics elements and adhered to PHE guidance (Fig. 1). We convened a workshop to refine the SMS and develop a pre-recorded voice message with an African economist and representatives from the University of Botswana, and Peek Vision’s Botswana office. Further iterations were made following a robust refinement process (Additional file 1: Appendix 1) that included input from laypeople, and professional translation and back-translation. The final messages are presented in Table 1.
Fig. 1
First draft SMS reminder. Note: ‘Tebogo’ and ‘Dr Dineo’ are not real names
We will use four arms as outlined below. Each SMS will be sent two times; on the day of referral and on the day before the appointment.
Initially, we will only test arms 1 and 2 (the control and intervention SMS messages). We plan to introduce the voice message arms after 6 weeks. This is because we are interested in introducing new arms at later stages in the screening programme and want to observe how the allocation algorithm handles the introduction of new arms part-way through testing established interventions.
Arm 1 (Control): Standard SMS reminder messages.
Arm 2: New SMS reminder messages.
Arm 3: Standard SMS reminder messages plus the pre-recorded voice reminder
Arm 4: New SMS reminder messages plus the pre-recorded voice reminder
Criteria for discontinuing or modifying allocated interventionsDue to the low-risk nature of the interventions, there will not be any formal option to discontinue or modify the reminder messages.
Strategies to improve adherence to interventionsThere are no relevant strategies to improve adherence. This is a pragmatic intention-to-treat study, and we will not collect data on whether messages were actually read or listened to by the intended recipients. A potential limitation of this study is that we cannot ensure that the message is actually delivered to- and read by the correct person. To an extent, this is true of all forms of phone-based reminder messages, as it is of paper reminder letters or notes sent by post, or home with children.
Relevant concomitant care permitted or prohibited during the trialNo other reminder messages will be sent from the Peek platform during the trial.
Provisions for post-trial careAs this is a negligible risk trial, no provisions will be made for post-trial care.
OutcomesAll children who are screened and found to need further assessment and treatment (e.g. refractive services) will be given an appointment, approximately 1 week later, at a specified field ‘triage and treatment’ clinic or at a specialist ophthalmic hospital clinic. The primary outcome is attendance at this pre-specified appointment on the appointment date (yes/no).
The Peek software retains a record of every referred child. When children attend for these appointments, they are checked in using Peek software. This automatically updates their attendance status. Attendance data will be automatically reviewed by an algorithm every 24 h. The great advantage of the Peek-based screening programme is that is a closed data system with complete, unified data records for every person screened, their referral status and their attendance status. No additional data collection activities are required.
Primary outcome: attendance at clinic on invited date. This is a binary outcome measure (yes/no). We will compare mean outcome rates between arms.
Secondary outcome: days elapsed between appointment date and attendance. This is because children may miss their appointed day but attend at a later date. We will compare mean number of days elapsed between each arm.
Subgroup analyses: attendance by age, sex, urban/rural residence, distance to clinic, ethnicity, guardianship, religion, language, household composition, migrant status, parental occupation, housing, assets and income.
Participant timelineThis automated adaptive trial will run continually for three working months (i.e. pausing during the school holidays when screening does not happen), recruiting participants until sufficient evidence has been gathered to reject the null (by triggering a stopping rule). Enrolment is planned to commence in quarter 3 2022. We intend to start with two SMS arms and add in voice messaging once the trial is underway. This is because we want to observe how the automated allocation system handles the introduction of new arms.
Sample sizeApproximately 1000 children will be screened every day. Based on previous programmes, we expect approximately 160 of these children to be identified as requiring referral for further assessment and treatment. All of these children’s parents/guardians would receive the standard SMS reminders in a standard programme.
The adaptive allocation method that we are using does not use a pre-specified a sample size. Instead, the study will run until one of two criteria is met:
Depending on the effect of the interventions, one of the stopping criteria might be met after a few days; however, it could also take years before reaching a definitive conclusion. We will set a 3-month limit for this current study due to resource constraints.
RecruitmentCommunity sensitisation is being led by the Ministry of Health and Ministry of Education. This includes TV and radio coverage explaining the Pono Yame screening programme. Our field coordinator will visit each region and work with schools to ensure that they are set up to enrol as many children as possible. Every referred child’s data will be included in the primary analysis. Subgroup analyses will only be permitted for children whose parents have consented for their sociodemographic data to be used for research purposes. This is a separate consenting process led by Peek.
Assignment of interventions: allocationSequence generation, concealment and implementationParticipants will initially be randomly allocated into two arms using computer-generated blocks of 12. As allocation and intervention delivery (sending SMS messages) is fully automated, there is no need for any of the human investigators to know participant allocation status. Once the first participants attend refractive services, the algorithm will begin adjusting the allocation ratio to favour the best-performing arms. There is no need for the investigators to see allocation status at this stage either. The data safety monitoring committee will be fully unmasked to allocation status and all outcome data and will have the power to stop the trial or suspend any arm.
Assignment of interventions: blindingWho will be blindedTrial participants will not be blinded. Programme implementers will check in participants when they attend clinic using Peek Capture. The software will automatically record the date and the time elapsed since referral. The adaptive algorithm will analyse attendance rates between arms according to pre-defined rules. Screening programme staff and data analysts will be blinded to assignment status. A small team of unblinded human statisticians will monitor the algorithm’s performance. They will double-check the algorithm’s working every 24 h during the trial and will repeat the final analysis comparing each arm. They will have the power to stop the trial, but they will not influence allocation.
Procedure for unblinding if neededThere is no procedure for unblinding.
Data collection and managementPlans for assessment and collection of outcomesReferral status, attendance status and days elapsed since referral will be collected using the Peek Capture system on Android devices. Every time a participant is referred and every time they attend at clinic, they are checked in using an android device operating Peek Capture software. Additional data on sociodemographic characteristics will be collected when participants initially present to the screening programme.
Plans to promote participant retention and complete follow-upAs the intervention is an SMS sent automatically by the programme, there is no scope for deviation. Similarly ‘loss to follow-up’ is the reciprocal for our primary outcome (attendance on appointed day).
Data managementData will be collected by Peek’s implementing partners using Android devices through the Peek Capture application. Peek Capture enforces security controls that include strong device passcodes and native Android encryption. Data stored is time limited, the device syncs via an encrypted connection with a Peek-managed server, the data is then deleted to minimise the risk of data stored on the device.
Data will be stored on a Peek-managed server hosted in a Virtual Private Cloud (VPC) utilising the Amazon Web Services (AWS) Cloud. Each Peek-powered programme is hosted on its own dedicated server and a VPC that will reside in the UK/EU ensuring all of the data privacy safeguards as governed under the GDPR. All data collected is securely stored in AWS data centres which are state of the art, utilising innovative architectural and engineering approaches. Routine manual data cleaning will be conducted periodically by Peek administrators. Internal software guardrails will pick up simple errors.
Data collected can be monitored using Peek Admin; it tracks the Programme progress, provides insights and helps ensure no one is left behind. Data exported from Peek Admin will be pseudo-anonymised removing names and any other key identifiers, only the least amount of data will be shared, and where possible it will be fully anonymised and aggregated for research purposes.
At the analysis stage, data will be sent via a secure file transfer, using an encrypted zip file to LSHTM researchers to perform statistical testing. The zip file will be saved on the protected LSHTM server and only authorised named project staff will be given access. Passwords will be sent separately. Further details can be found in the Data Management Plan (Additional file 1: Appendix 2).
ConfidentialityPeek routinely collects sociodemographic information from each child who is referred on for refractive services including age, sex, location, ethnicity, religion, parents' occupation, parents' education, housing characteristics and asset ownership. This information will be held on a Peek-managed server hosted in a Virtual Private Cloud (VPC) utilising the Amazon Web Services (AWS) Cloud. Peek also seeks consent to use this data for research purposes.
Sociodemographic data on participants who have provided consent will be shared with the statistical analysis team at LSHTM for subgroup analysis. All team members who will access these data will have undertaken information security training. We will use encrypted data transfer and avoid cloud services outside the EU. The aggregated Peek data that is shared with LSHTM project staff will not contain any names; however, the data being shared may still permit the identification of individuals depending on the domains being shared and may therefore constitute pseudo-anonymised data. All data arising from this project will be stored securely for 10 years. Further information is provided in the data management plan (Additional file 1: Appendix 2).
Plans for collection, laboratory evaluation and storage of biological specimens for genetic or molecular analysis in this trial/future useNot applicable. We will not be using biological specimens.
Statistical methodsStatistical methods for primary and secondary outcomesThis study will use Thompson sampling a Bayesian approach to identify the best arm. This is a Bayesian algorithm widely used to learn about arms and optimise decision making [27] Every 24 h, the probability of each arm being the best arm overall will be estimated, using Monte-Carlo simulations to get the posterior probability estimates. As there is no evidence available on how the messages would perform relative to another, a regularising prior of Beta(100,000) (i.e. centred at p=0.5 with a 90% credible interval of 0.44–0.56) will be used to avoid overfitting extreme data in the early phase of the trial. It is expected that about 1,000 children will enrol every day, and the observed data will begin to dominate the prior within the first couple of days. Each arm will have a probability of being best between 0 and 100%, and the sum of all two probabilities will equal 100%. These probabilities will be compared to the stopping rules as to whether the trial should stop or continue into the next day. If the trial is to continue, the proportion allocated to each arm for the next day will be updated to be proportional to the estimated probabilities. We will conduct all analyses using the runif and rbeta functions in R (R Foundation for Statistical Computing, Vienna, Austria). Figure 2 illustrates participant flow and operation of the algorithm.
Fig. 2
Interaction between patient flow and the adaptive trial algorithm
Interim analysesThis is an automated adaptive trial. Our algorithm will review the attendance data every 24 h and perform statistical testing. Two stopping rules will be applied during these daily interim analyses:
1.There is a >95% probability that one arm is best.
2.There is a >95% probability that the difference between the arms remaining in the study is <1%.
If neither of these rules have been satisfied, then the trial (i.e. enrolment) will continue until three months of active screening have elapsed. The Bayesian algorithm will adjust the allocation ratio based on the performance of each arm with respect to the updated posterior probability that each is associated with attendance (Fig. 3).
Fig. 3 Methods for additional analyses (e.g. subgroup analyses)
Methods for additional analyses (e.g. subgroup analyses) Internal data from a pilot site suggests that around 10% of children who attend the ‘triage and treatment’ clinic will subsequently be identified as having an eye need that requires further specialist ophthalmological assessment in a hospital clinic. These children will be referred from the ‘triage and treatment’ clinic to the local hospital. This subgroup will also receive either the intervention or control reminder messages. Again, the outcome will be attendance on appointed date.
Once the trial is complete, we will perform retrospective subgroup analyses to explore whether attendance within each group was associated with sociodemographic variables. We use multivariable logistic regression to assess whether each sociodemographic variable is associated with attendance. We note that this is an exploratory analysis, providing hypotheses that can be tested in subsequent studies.
We will perform a secondary analysis that excludes participants whose preferred language is neither Setswana nor English.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing dataThe primary analysis only requires trial arm and the outcome (attendance) to be recorded. The trial arm should be recorded automatically as part of the Peek coding, and if it was missing it would be due to a bug in the coding. If this occurred, there is no statistical method that could be used to recover that data so any records with trial arm missing would not be included in the updating of the probability that an arm is best. We will check the code every 24 h to ensure that it is running as expected and correct any errors that we find immediately. The outcome cannot be missing, as a participant is set as ‘not attended’ until the point where they are updated as having ‘attended’.
Plans to give access to the full protocol, participant-level data and statistical codeThe full protocol is available from the corresponding author. Statistical code will be made freely available online using GitHub. In line with the UK concordat on open research data (2016), anonymised participant-level data from this trial will be made available to bona fide research groups (evidenced via curriculum vitae and the involvement of a qualified statistician), and in line with the trial’s publicly available data sharing policy, following review and approval from the trial’s data monitoring committee. No reasonable request will be turned down, and the appropriate data will be made available within 1-month of receiving the request. There may be multiple levels of permission required in-country before data can be shared, including national ministry of health approval and local implementation partner approval.
Patient and public involvementLaypeople were involved in checking the wording of the intervention messages and suggesting refinements that better conveyed their underlying meaning.
Oversight and monitoringComposition of the coordinating centre and trial steering committeeTrial coordinating centre:
Dr Luke Allen, Co-Principle Investigator and trial manager, LSHTM
Hannah Chroston, lead administrator, LSHTM
Bakgaki Ratshaa, trial coordinator, University of Botswana
Trial management group
Prof Andrew Bastawrous, chief investigator
Prof Oathokwa Nkomazana, co-PI
Dr Luke Allen, co-PI
Prof Matthew Burton, methods advisor
Dr David Macleod, lead statistician
Dr Nigel Bolster, Peek integration
Min Kim, statistician
Dr Ari Ho-Foster
Dr Michael Gichangi, methods advisor
Data management team
Dr Luke Allen, co-PI
Dr David Macleod, lead statistician
Dr Nigel Bolster, Peek integration
Min Kim, statistician
Composition of the data monitoring committee, its role and reporting structureAn independent Data and Safety Monitoring Board (DSMB) will be appointed by the trial steering committee. The DSMB will have three members, all independent of the running of the trial with relevant clinical and epidemiological experience.
The DSMB will confirm their specific meeting arrangements. It is proposed that the DSMB would meet prior to the beginning of the trial (Q2 2022), one third of the way through, and at the end, to assess the safety of the trial procedures. The DSMB will agree the way it will monitor the data, what it requires from the investigators in this respect and will communicate this to the PIs. All data can be interrogated remotely in real time.
The DSMB may visit the study coordination centre to assess data management, record keeping and other important activities. The DSMB will determine the manner in which it will monitor the data, what it requires from the investigators in this respect and will communicate this to the PIs.
Adverse event reporting and harms Definitions Term Definition Adverse event (AE) Any untoward medical occurrence in a patient or study participant Serious adverse event (SAE) A serious event is any untoward medical occurrence that:All adverse events will be reported. Depending on the nature of the event the reporting procedures below will be followed. Any questions concerning adverse event reporting will be directed to the study coordination centre in the first instance. The flow chart below has been provided to aid the reporting of AEs.
Responsible personnel Chief Investigator (CI)The CI has overall responsibility for the conduct of the study and the ongoing safety and evaluation of any IMPs being used in the trial.
Promptly notifying all investigators, Institutional Review Board (IRB) or Independent Ethics Committee (IEC) and Competent Authorities (CAs) of each concerned member state of any findings that may affect the health of the trial participants.
Keeping detailed written reports of all AEs/ARs identified in the protocol as critical to the evaluation of safety within the agreed timeframes specified in the protocol.
Accurate production and submission of the Development Safety Update Reports and progress reports to CAs and IRB/IECs.
Collate all AR/AEs/SAEs/SARs and report to the Sponsor annually.
Ensure that the PIs report all SAEs/SUSARs immediately to the Sponsor and to the CAs, IRB/IECs and any other relevant parties within agreed timelines
Supplying the Sponsor and IRB/IEC with any supplementary information they request.
Principal Investigators (PI)The PIs have responsibility for the research performed at the local site, handling and management of investigational medical products, and informing the CI, Sponsor, Ethics, regulatory bodies and the trial coordinating team, of all adverse events that occur at their site
Safety responsibilities:
Ensure trial participant safety and the swift and adequate management of trial participants with any type of AE/AR as per the management protocol described below.
Reporting all SAEs/SUSARs immediately to the Sponsor and to the CAs, IRB/IECs and any other relevant parties within agreed timelines (i.e. LSHTM, EFMHACA, ORHB, FMOST).
Assessing each event for causality, severity and expectedness. (Note: a medical decision which must be made by the investigator directly involved with the care of the patient/participant experiencing the AE)
Ensure adequate archiving of AE records and reports in the local trial office along with the trial master files.
Collate all AR/AEs/SAEs/SARs biannually and present to the CI.
Guide and supervise the field research team on accurate recording, reporting of all adverse events.
Field Research Team Members (Coordinators, Nurses, Examiners, Recorders)All field research team members are responsible for identifying, recording and reporting any AE or AR to the PIs regardless of severity or causality.
Assessing each event for causality, severity and expectedness. (Note: a medical decision which must be made by the investigator directly involved with the care of the patient/participant experiencing the AE).
Ensure that the participant has received the necessary management. This includes advice/reassuring, referral, offering transport, paying for management, making follow-up visits
Report to the PIs/Project manager AEs/ARs based on the specified timeline and file all AE/AR recorded forms in the trial master file.
Non-serious AEsAll non-serious AEs will be reported to the study coordination centre and recorded in a dedicated AE log within 72 h. The entry must state the patient ID, date and time of AE, nature and relation to the intervention, if any. The AE should also be reported to the data and safety monitoring committee within 72 h. AE logs will be stored on a secure, password-protected file on a LSHTM computer.
Serious AEsSerious adverse events (SAEs) will be reported to the PI and study coordination centre within 24 h of the local site being made aware of the event. The PI will report the event to the data safety monitoring committee within 48 h and include it in the study safety report.
An SAE form will be completed and submitted to the PA and study coordination centre with details of the nature of event, date of onset, severity, corrective therapies given, outcome and causality. All SAEs whether expected, suspected or unexpected will be reported to regulatory bodies and the trial DSMB within 48 h of occurrence. The responsible investigator will assign the causality of the event. All investigators will be informed of all SAEs occurring throughout the study. If awaiting further details, a follow-up SAE report should be submitted promptly upon receipt of any outstanding information.
Any events relating to a pre-existing condition or any planned hospitalisations for elective treatment of a pre-existing condition will not need to be reported as SAEs.

Contact details for reporting SAEs
Please send SAE forms to: luke.allen@lshtm.ac.uk or nkomazanao@UB.AC.BW using the title ‘SAE’
Tel: +44 (0) 20 7958 8316 (Mon to Fri 09.00–17.00)
Tel: +267 355 0000
Frequency and plans for auditing trial conductThe study may be subject audit by the London School of Hygiene & Tropical Medicine under their remit as sponsor, the Study Coordination Centre and other regulatory bodies to ensure adherence to Good Clinical Practice.
Plans for communicating important protocol amendments to relevant parties (e.g. trial participants, ethical committees)Any important protocol modifications will be reported to the co-investigators, research committees, the trial registry and—where appropriate—journals and regulators via email.
Dissemination plansScientific results will be published in Open Access in peer-reviewed journals and presented at relevant international conferences. All publications and presentations relating to the study will be authorised by the Trial Management Group. The first publication of the trial results will be in the name of the Trial Management Group members. Members of the Data and Safety Monitoring Board will be listed and contributors will be cited by name if published in a journal where this does not conflict with the journal’s policy. Authorship of any parallel studies initiated outside of the Trial Management Group will be according to the individuals involved in the project but must acknowledge the contribution of the Trial Management Group and the Trial Coordinating Centre.
留言 (0)