
記住我
Besides screening and diagnosis, one of the major needs related to the SARS-CoV-2 pandemic is to collect and analyze a considerable amount of viral genomes, to guarantee a rapid geographical and continuous surveillance of VOC. To achieve this goal, we developed a systematic workflow that allows the collection, whole genome sequencing (WGS), cloud data processing, and sharing of up to 4500 SARS-CoV-2 genomes per week. Our approach is based on the optimization of an amplicon-based workflow [16] (see the “Methods” section) (Fig. 1A). To both increase processivity and efficiently reduce costs, the protocol was tested and validated with a decreasing amount of input RNA for the generation of the libraries. In particular, we tested 5 μL, 2,5 μL, 1,25 μL of unquantified RNA and proportionally scaled down the reaction volumes to 1/2, 1/4, and 1/8 (solution A, B, and C, respectively) (Fig. 1B, C, Additional file 1: Fig. S1A and Additional file 2: Table S1). Neither the number of mapping reads, the genome coverage, nor the number of sequences passing our quality filters and submitted to GISAID were significantly affected by volume reductions.
Fig. 1
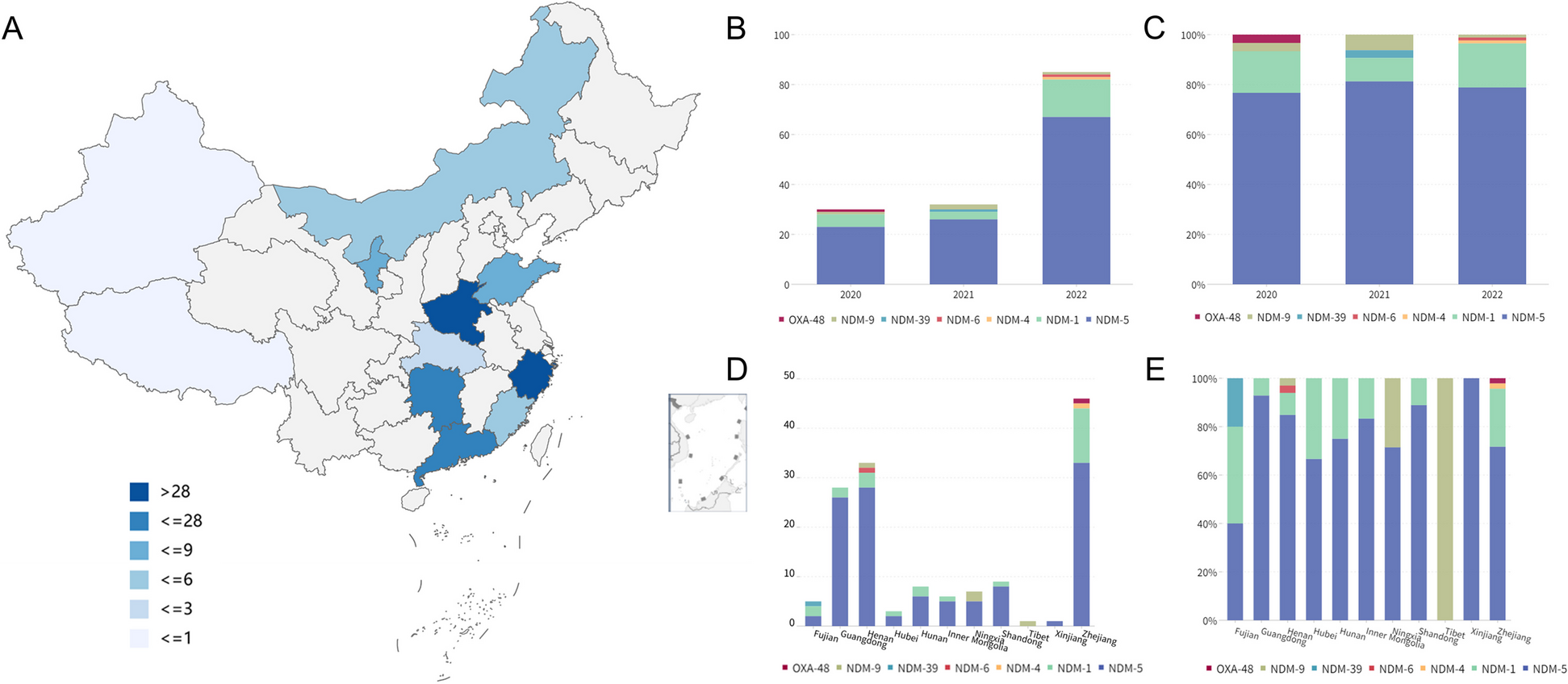
A systematic approach allows the generation of large and robust genomic data in a cost-effective manner. A Schematic representation of the workflow set up to collect, process, and analyze a considerable number of viral genomes. Top: Oronasopharyngeal swabs are performed to diagnose the presence of the SARS-CoV-2 genome in patients and extract its RNA. Subsequently, viral RNA is retrotranscribed and subjected to two PCR steps to amplify and index the obtained cDNA. After circularization and nanoball generation, the obtained library is then sequenced and analyzed. Bottom: As an alternative and faster approach, an optimized approach enables the amplification and indexing to occur in one PCR step. B Multiple solutions were tested to optimize the workflow. The table reports the input RNA volume, the amount of reads produced per sample, the number of samples loaded per flowcell, the average time required to process a 96-well plate, and the relative cost per sample. Cost details are reported in Additional file 2: table S1. C Boxplot showing the percentage of samples submitted on the GISAID platform, divided by each tested solution. Only samples with an average Ct value < 33 were considered. D Violin plot showing the distribution of the percentage of SARS-CoV-2 reads detected for different ranges of CTs. n:sample size. E Variant annotation, cumulative frequency, and sequencing coverage of each position of the SARS-CoV-2 genome. F Venn diagram showing the intersection between mutations detected in all the sequenced genomes worldwide (yellow) and the mutations found in this study (light blue). G Representation of all the 156 lineages identified in this study. The length of the bars is indicative of the number of samples for each lineage in the logarithmic scale. Colored bars indicate VOC
Being able to rapidly process the RNA sample to the final viral genome consensus is a critical aspect for retrieving meaningful data on the SARS-CoV-2 genome surveillance in a territory. We addressed this point by both optimizing the steps required for library generation and adjusting the number of samples sequenced in each run (Fig. 1B). Particularly, we merged the targeted and the indexing amplification steps in a single PCR (Fig. 1A, see the “Methods” section). Such a one-step strategy has an efficiency of ~ 60%; however, it allows to remove a magnetic beads purification step and thus reducing the hands-on time for library generation by ~40% (Fig. 1B). In parallel, sequencing flow performance and efficacy were tested and validated against increasing number of samples per run, as such can have an adverse effect on the quality of the resulting viral sequences. We compared QC statistics obtained by sequencing at a depth of ~ 9, 4.5, and 2.75 million 100bp paired-end reads per sample. This translates into sequencing two (192 samples), four (384 samples) or eight (768 samples) 96-well plates per run. As expected, the total amount of reads decreases by increasing the number of samples per flow-cell; nevertheless, the downstream parameters were not affected, allowing us to genotype a similar amount of SARS-CoV-2 genomes (i.e., GISAID database sequence acceptance rate—Fig. 1C).
Therefore, by finely tuning the starting amount of RNA, the library generation steps, and the number of samples loaded in each sequencing run, we were able to decrease both the processing time and the costs. Using our optimized SARS-CoV-2 WGS workflow solution B, during 2021, we were able to process, as a proof-of-principle approach, about 30,000 swabs and sequence 22,228 of them, 17,193 of which generated high-quality genomes (defined as those complete genomes with a percentage of Ns lower than 5%, Additional file 2: Table S2). A strong correlation between the number of reads detected in each sample and the Ct values obtained from a diagnostic qPCR was observed (Fig. 1D). SARS-CoV-2 reads showed a proportional rate with respect to Ct in the intervals between 40 and 25 Ct while reaching saturation <25 Ct. Altogether, these observations suggest that our WGS approach reliably quantifies the viral load and provides us with crucial metadata to correlate higher virus titer to specific virus lineages and a transcriptional response from host cells (see later in Figs. 2 and 4). The robustness of this approach was further established by analyzing the mean coverage level, which appeared to be homogeneous across the entire sequence (Fig. 1E). This piece of evidence confirmed the absence of major biases in the single nucleotide evaluation: hence, we investigated the SNPs information derived from our genomic screening and determined missense and synonymous mutations to be the most frequent across the entire genome, although few positions appeared to be more prone to mutate (Fig. 1E). Indeed, a number of mutations (Additional file 2: Table S3) were efficiently detected in our dataset, 194 of which were previously unknown (Additional file 2: Table S4) and 40 only identified in Campania (Fig. 1F). Interestingly, out of the 194 mutations first collected in the region, 20 fall within the Spike gene, at or nearby residues reported to be relevant for neutralizing antibody binding [28]. Taken altogether the mutations detected allowed us to identify 156 different SARS-CoV-2 pangolin [6] lineages (Fig. 1G), some of which were retrieved for the first time thanks to our activity (see below).
Fig. 2
Characterization of SARS-CoV-2 genome evolution in the south of Italy. A Geographic map representing European States, colored by the number of 2021 months with at least 5% of viral genomes sequenced, compared to new cases. Only for Italy, individual regions are displayed. B Top: geographic map representing Italian regions, colored by the number of genomes deposited on the GISAID platform. Bottom: percentage of genomes deposited on GISAID over the total Italian sequences, divided in Northern (green) and Southern (blue) regions. 28% of the overall Italian sequences have been produced by this study (dark blue). C Geographic distribution in Campania of the genomes analyzed in this study (top) relative to the population density (bottom). D Density plots showing the distribution, in time, of the most frequent variants described in this study (middle) or in Italy (bottom) relative to the Campania infection curve (top) and waves (red-colored areas). Red arrows highlight different variants dynamics between regional and national level, in a certain period of time. E Distribution of the average CT value across different Variants of Concern (VOC). Only not significant (n.s.) pairwise comparisons are reported (Bonferroni adjusted p-value > 0.05)
Characterization of SARS-CoV-2 genome evolution in the south of ItalyAs aforementioned, starting from the end of December 2020 to the first week of 2022, we have sequenced, uploaded to GISAID, and analyzed 17,193 SARS-CoV-2 genomes. Our workflow was tested throughout the Campania region, which includes the major southern Italian metropolitan areas and some of the most densely inhabited cities in Europe. Globally, we were able to sequence, in most months during 2021, at least 5% of all COVID-19 positive samples (Fig. 2A), making Campania compliant with EC/ECDC recommendations, reaching a sequencing coverage comparable to that of North-European countries. In fact, our dataset represents almost half of all sequences retrieved and uploaded from the south of Italy and 28% of all sequences produced in the country (Fig. 2B). Samples were collected starting from March 2020, by randomly selecting positive swabs and reflecting population demographics of sex, age, and the geographical distribution across the area of interest (Additional file 1: Fig. S2A and Fig. 2C).
The analysis of samples collected during the whole pandemic period allowed us to unveil the full dynamics of the SARS-CoV-2 outbreak in Campania. We indeed reconstructed the distribution of all the VOC arrived in Campania (Additional file 1: Fig. S2B), particularly the delta (represented by B.1.617.2 and AY.* lineages) and alpha (B.1.1.7 and Q.*) VOC, which represented the vast majority of variants detected (71.6%). In accordance with worldwide data, the first VOC arriving in the region, starting from December 2020, were the B.1.1.7 and P.1 (gamma variant). Next, other VOC were detected in the region, including B.1.351, P.1, and B.1.1.529 lineages (i.e., beta, gamma, and omicron VOC, respectively). We also identified three main Variants of Interest (VOI); the B.1.427, B.1.525, and B.1.621 lineages (i.e., epsilon, eta, mu).
Out of the 156 viral lineages identified in the region, 5 were first recorded in Campania territory, namely B.1.1.187, B.1.177.33, B.1.177.75, C.18, and P.1.1 (Additional file 2: Table S5). In particular, the C.18 viral variant was first collected in July 2020, whereas its first record outside Campania was registered 3 months later, suggesting a possible epidemiological origin from our territory of investigation. Similarly, over 82% of B.1.1.187 samples collected during the pandemics were derived from Italy, all from Campania. Our analysis also showed that the first gamma VOC sub-variant identified (pangolin lineage P.1.1) was first sampled in Campania by our activity (Additional file 2: Table S6) and that it was specifically enriched in Italy, with sequences from Campania representing about 20% of all P.1.1 samples identified.
Looking at the whole picture, we determined that the main infection peaks in the region were associated with the spread of specific viral lineages (Fig. 2D top and middle overlays). Indeed, while the first wave of infections was mostly due to the ancestral B.1 lineage, the second one (autumn 2020) was led by the B.1.177 lineage (also referred to as the European or Spanish variant) and its sub-lineages. Interestingly, the time window between the first two infection peaks was characterized by two of the aforementioned lineages to be firstly detected in Campania; B.1.1.187 and C.18, associated with most of the COVID-19 cases during late spring and summer of 2020. These two variants distinguish the pandemics in Campania relative to the rest of Italy (Fig. 2D lower level, red arrow). In the same period in the rest of the country, infections were predominantly associated with other B.1 sub-lineages, mainly including B.1.1, B.1.1.305, and B.1.1.229.
Finally, the two infection peaks of 2021 were due to the spread of alpha and delta variants. These two VOC succeeded one after the other and accounted for almost all COVID-19 cases in the first (alpha) and second (delta) half of 2021. Interestingly, from December 2021 B.1.1.529 (omicron) variants started to emerge.
Since during this succession of variants in the regional territory, none of them ever reappears after being undermined by the subsequent one, it is fair to suppose that each variant has been substituted by one with higher fitness and capability to spread. To test this hypothesis, we looked at the viral loads in the upper respiratory ways of patients infected by the predominant variants in Campania (Fig. 2E). We observed a clear trend towards an increase of viral titer in patients during the pandemics, with a Ct value difference between omicron and the ancestral B.1 variant of −7.8 (q value < 2×10−16 pairwise Mann-Whitney test). A similar trend towards decreasing Ct values was observed also when taking into account all the variants identified in the region (Mann-Kendall test, p value=8.99×10-10, Additional file 1: Fig. S2C).
High-throughput genomic surveillance allows the identification of new variants based on the analysis of single mutationsAs, the comparative analysis of our dataset with GISAID world data allowed us to retrospectively identify viral variants firstly sampled in Campania (B.1.1.187 and C.18), we were interested to explore whether it was possible to unveil new viral lineages circulating in the territory. To achieve this goal, we explored several approaches. We mainly focused on the concept that a new SARS-CoV-2 variant is characterized by a specific set of mutations, therefore we generated approaches based on (1) mutations associated with a higher infectivity found in unexpected variants; (2) an increasing incidence of a set of mutations in a short time window; (3) the appearance of new mutations in samples collected by patients with persistent infections.
First, we explored SARS-CoV-2 “mutations of concern” genotyped in unexpected lineages (Fig. 3A and Additional file 1: Fig. S3A). Interestingly, we found that the Spike E484K substitution had an unexpected distribution in the lineage identified at the beginning of 2021. This mutation is typically found in P.1.x and B.1.351.x viral lineages and has been associated with a decreased sensibility to both monoclonal and BNT162b2 vaccine-induced antibodies [7, 10, 11, 29]. However, as of May 2021, ~21% carrying this mutation were associated with the B.1.177.x lineage. To further investigate this finding, we performed a phylogenetic analysis over our entire dataset using Nextstrain [19] and found that all B.1.177.x samples carrying the Spike E484K substitution (B.1.177E484K samples) clustered in a specific and monophyletic clade branching within the B.1.177.x lineage (Fig. 3B). We further confirmed this finding by looking at the distribution of B.1.177E484K samples in the phylogenetic tree containing all high-quality SARS-CoV-2 genomes from GISAID [6, 30]. This data points to the fact that B.1.177E484K samples cluster in a monophyletic clade with an extremely high (0.99) support value, thus confirming regional level incidence (Additional file 1: Fig. S3B). Additionally, as the GISAID database revealed that B.1.177E484K samples had been identified for the first time in Campania through our program, we investigated their geographic distribution in the regional territory to trace the epidemiological link (Fig. 3C). Surprisingly, these samples originated from a specific area between Naples and Salerno called “Agro Nocerino-Sarnese.” Combining these results, we hypothesized that B.1.177E484K variant had probably arisen in this area in December (treetime divergence inferred interval: 2020-11-22~2020-12-21) and, then, spread nearby in Campania and in other confining Italian regions (mainly Lazio and Basilicata). Altogether, these observations allowed us to define a new SARS-CoV-2 lineage, which is now recognized by the Pangolin nomenclature B.1.177.88.
Fig. 3
High-Throughput genomic surveillance allows the identification of new SARS-CoV-2 lineages. A Donut chart representing the amount of analyzed genomes presenting the Spike E484K mutation, divided by lineage. The definition of Expected lineage is described in the Methods. B Section of the phylogenetic tree representation of the whole dataset (n=12,998), colored by lineages. The identified lineage is reported (blue dots, left) and zoomed in (right). n:sample size. C Geographic distribution of genomic variants belonging to the identified lineage, colored by the collection date. The size of each pie chart is proportional to the number of samples in each geographic position. n:sample size. D Line plot showing the frequency trend of the selected mutations in time. E Section of the phylogenetic tree representation of the whole dataset (n=12,998), colored by lineages. The identified lineage is reported (arrow, blue dots). n:sample size. F Geographic distribution of genomic variants belonging to the identified lineage, colored by the collection date. The size of each pie chart is proportional to the number of samples in each geographic position. n:sample size. G Genomic characterization of twenty patients with long COVID-19 infection. The number of detected mutations is reported as a function of the number of days from the first swab. The assigned lineage (colors) and consistency (transparency) are also displayed. H Patient 8 genomic characterization relative to the number of detected mutations (colors), the infection load (y-axis), and symptoms severity (+++: severe; ++: moderate)
To identify any new variant rapidly growing in the territory as soon as it appears, we exploited another approach based on the incidence over time of each of the 6441 amino acid mutations we identified. As, by definition, a viral variant is defined by a specific combination of mutations, we looked at mutations that displayed similar trends in the same period and grouped them in clusters. In order to identify any potential new alpha subvariant growing in Campania, we applied this methodology to the data collected until may 2021, when the variant reached its maximum (Additional file 1: Fig. S3C). Several clusters clearly reflected the trends of known lineages, confirming the robustness of our approach; for instance, cluster 6 consisted of those substitutions that characterize the B.1.177.x lineage (namely N A220V and Spike A222V) and presented the same trend over time. Similarly, cluster 29 reflected the trend of B.1.1.7 lineage. Among these, cluster 18 was particularly interesting (Fig. 3D). It consisted of 4 mutations (NSP2 Y316C, NSP3_T1306I, NS7a T120I, NS8 H112Y) with the exact same frequency behavior over time, thus suggesting a possible SARS-CoV-2 haplotype. A further investigation revealed that these SNPs define a set of samples assigned to the B.1.1.7 lineage and specifically localized in Campania in May 2021 (B.1.1.7YTTH samples). Similarly to the previous analysis, we carried out a phylogenetic analysis that confirmed B.1.1.7YTTH as monophyletic (Fig. 3E and Additional file 1: Fig. S3D). While B.1.1.7YTTH genomes did not show any geographic enrichment, its temporal distribution was indicative of an inland origin (Treetime divergence inferred interval: 2020-12-01~2020-12-06), followed by its spread first to the Neapolitan coast and then towards the Southern Neapolitan province (Fig. 3F). B.1.1.7YTTH variant has been recognized, upon our alert, as one of the first B.1.1.7 sublineage by the Pangolin system and is now referred to as the Q.2 lineage.
By applying the same approach to the data produced till January 2022, we also identified a new Omicron subvariant (BA.1.21.1, Cluster 17 in Additional file 1: Fig. S3E and F). The variant is characterized by an early STOP codon mutation in NS7b (E3stop) and a SNP in Nsp12 (L749M). This variant was first collected at the end of 2021 and rapidly spread in Campania at the beginning during 2022, accounting for over 10% of all the infections in the region between January and March 2022.
Several reports [31] showed the accumulation of mutations in the SARS-CoV-2 genome during persistent infections. However, the frequency of such events is still overlooked. In order to possibly address this question and identify potential new variants, we analyzed swabs collected from 20 patients multiple times for over 40 days during prolonged infections (Fig. 3G). Age and immunological status highly varied across the patients (Table 1): patients’ age ranged from 13 to 88 (average 62) years and while most of them were affected by simple or bilateral pneumonia, six suffered a more severe respiratory failure and only one showed no COVID-related symptomatology. It is worth noting that, although most samples were collected during 2021, none of the patients had completed a three-dose SARS-CoV-2 vaccination cycle, 4 had only one vaccine dose, and most had no vaccination at all (13/20).
Table 1 Detailed clinical status of patients from Fig. 3GSequencing of the viral genetic material confirmed no shift from a viral variant to another over time but each had a set of patient-specific mutations. However, looking at the individual mutations, in one patient (#8) there was an actual increase in the number of amino acid substitutions, as confirmed by several independent sequencing runs on several subsequent timepoints. The acquisition of the mutation (NSP13 R339C) was recorded only after 40 days from the first swab and did not correlate with an increase in the viral load or a worsening of the symptoms (Fig. 3H).
These results suggest that in specific conditions, such as over 40 days of persistent infection, the SARS-CoV-2 genetic consensus sequence can actually change, although the rate of such an event, as well as its biological significance, are not known yet.
Tracking new variants based on mutations arising in specific conditions is a novel approach for SARS-COV-2 surveillance. Here we showed that, by combining this approach with deep profiling of viral variability, new SARS-CoV-2 variants can be unveiled, even at the regional level.
Transcriptional profiling of Sars-CoV-2-infected patients reveals a gene signature correlated with viral load and preserved across different lineagesThe comprehensive gene expression profiling of the respiratory epithelium of patients positive for SARS-CoV-2 infection holds great promises in terms of preventive, diagnostic, and therapeutic advancements. For this reason, we implemented an RNA-seq workflow adapted to work with diagnostic swabs, known to have extremely low quantity and quality of RNA. We processed around 700 samples, divided in two batches for the analysis of the differential molecular host response to B.1 and Delta variants infection (Additional file 1: Fig. S4A). After filtering, the B.1 final dataset comprehended 116 SARS-COV-2 positive samples to be compared with 88 negative ones. On the other hand, the Delta dataset was composed of 43 and 95 SARS-COV-2 positive and negative samples, respectively (Additional file 1: Fig. S4A). Although the cohort of patients was numerous, in both cases many confounding variables influenced the possibility to compare positive and negative conditions. Inter-patient heterogeneity, different viral loads, and swab-related variability are some of the factors that prevented us from finding a strong variance solely related to the presence or absence of the infection (Additional file 1: Fig. S4B). Therefore, we decided to take advantage, again, of the Ct values associated with positive samples and perform a correlation analysis between gene expression and viral load, starting with the B.1 dataset (Fig. 4A top). After filtering non-expressed genes (see Methods), a Pearson correlation test was conducted and a signature of 161 genes (Additional file 2: Table S7) was found to be significantly anti-correlated with Ct values (p-value < 0.0001, Fig. 4A bottom). Among the 10 most anti-correlated genes, many downstream targets of interferon antiviral response (e.g., IFI44L, OAS2, PARP9, IFITM3, IFIT1) were found, as already reported from in vitro experiments and single-cell studies [32]. We confirmed an enhanced antiviral immune response by performing pathway and gene signatures enrichment analyses (Fig. 4B). Indeed, together with COVID-19- and Bronchitis-related signatures, the most significant results comprehended Interferon Alpha pathway and its inducers, IRFs. Additionally, STAT3-regulated genes were enriched, which were recently found to be aberrantly activated upon SARS-CoV-2 infection [33] (Fig. 4B). Interestingly, when looking at the expression levels of these genes in our cohort, negative patients displayed a transcriptional behavior comparable to samples with the lowest viral load (Fig. 4C). We applied the same approach to the Delta dataset and retrieved a molecular signature of 16 genes (Fig. 4D and Additional file 1: Fig. S4C-E), way smaller than the other one, most probably due to the restricted number of patients. Nevertheless, almost every gene (13 genes, 81.25%) was common to the B.1 signature and belonged to the same pathways (IFIT3, OAS3, IFI6 - Fig. 4D). With that said, our CT-based approach overcomes all the technical and biological variability related to the direct use of regular swabs extracts and establishes a robust gene signature that is preserved across different viral lineages and could be used as biomarkers for disease monitoring, prevention, and non-conventional treatments.
Fig. 4
Transcriptional profiling of SARS-CoV-2 infected patients reveals a gene signature correlated with viral load and preserved across different lineages. A Correlation analysis between CTs and gene expression of B.1 patients, performed on 8100 genes, is shown as a barplot. For each gene (x-axis), its correlation value (y-axis) and significance (p-value < 0.0001, red) is reported. Bottom: highlight of the significant results. (161 genes). The top 10 most anti-correlated genes are reported (black box). B Pathway and gene set enrichment analysis performed for different databases using the gene signature previously identified. Each barplot shows the significance (x-axis) and the percentage of overlap (fill color) between the input signature and the tested public genesets. C Heatmap of z-scored, log2-transformed, and normalized gene counts for the 161 significantly correlated genes from A. Values have been averaged in 4 groups of samples depending on the CT (x-axis) or whether they were negative. D Venn diagram of significantly anti-correlated genes between B.1 (161 genes) and Delta (16 genes) variant-infected patients
留言 (0)