
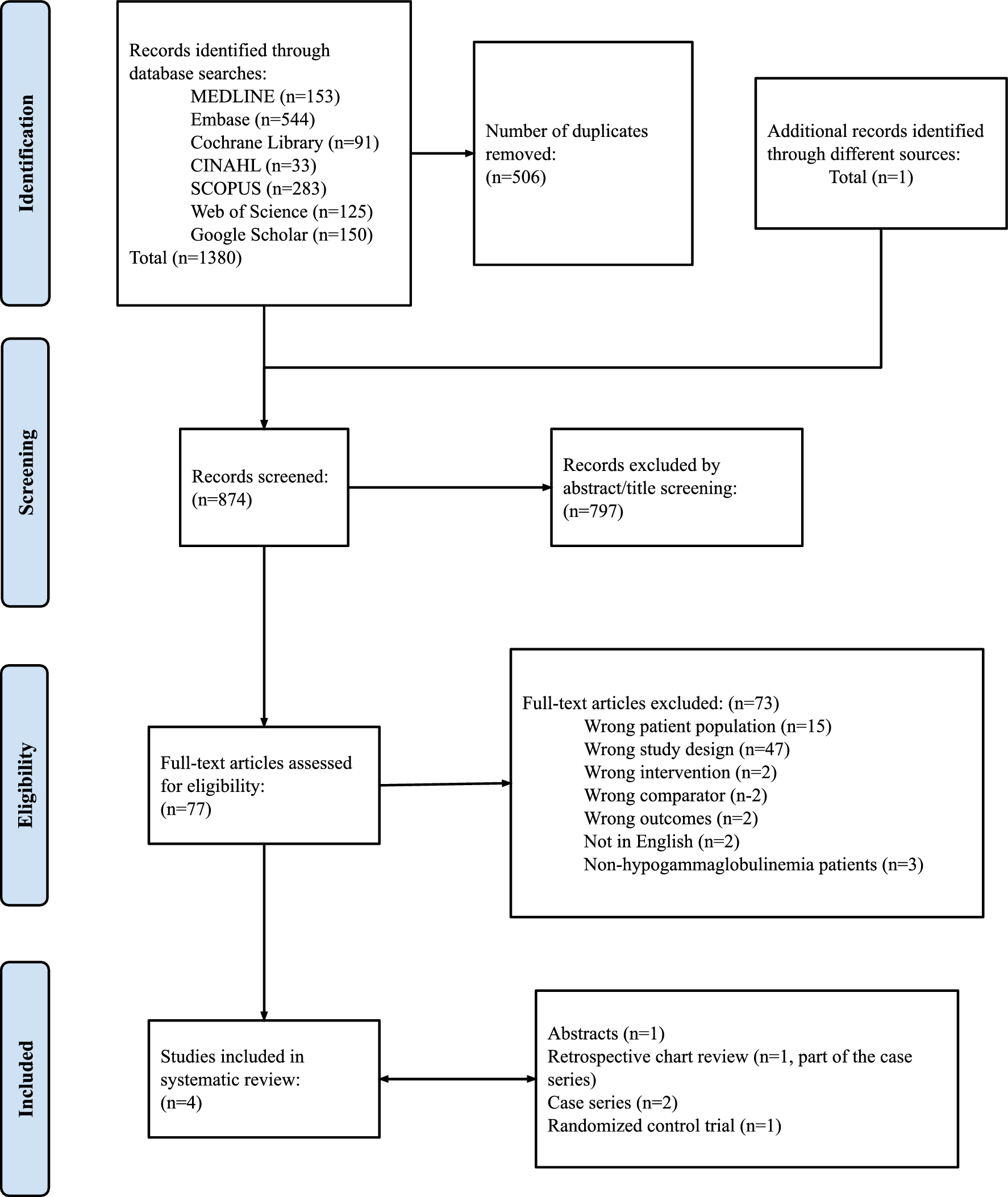
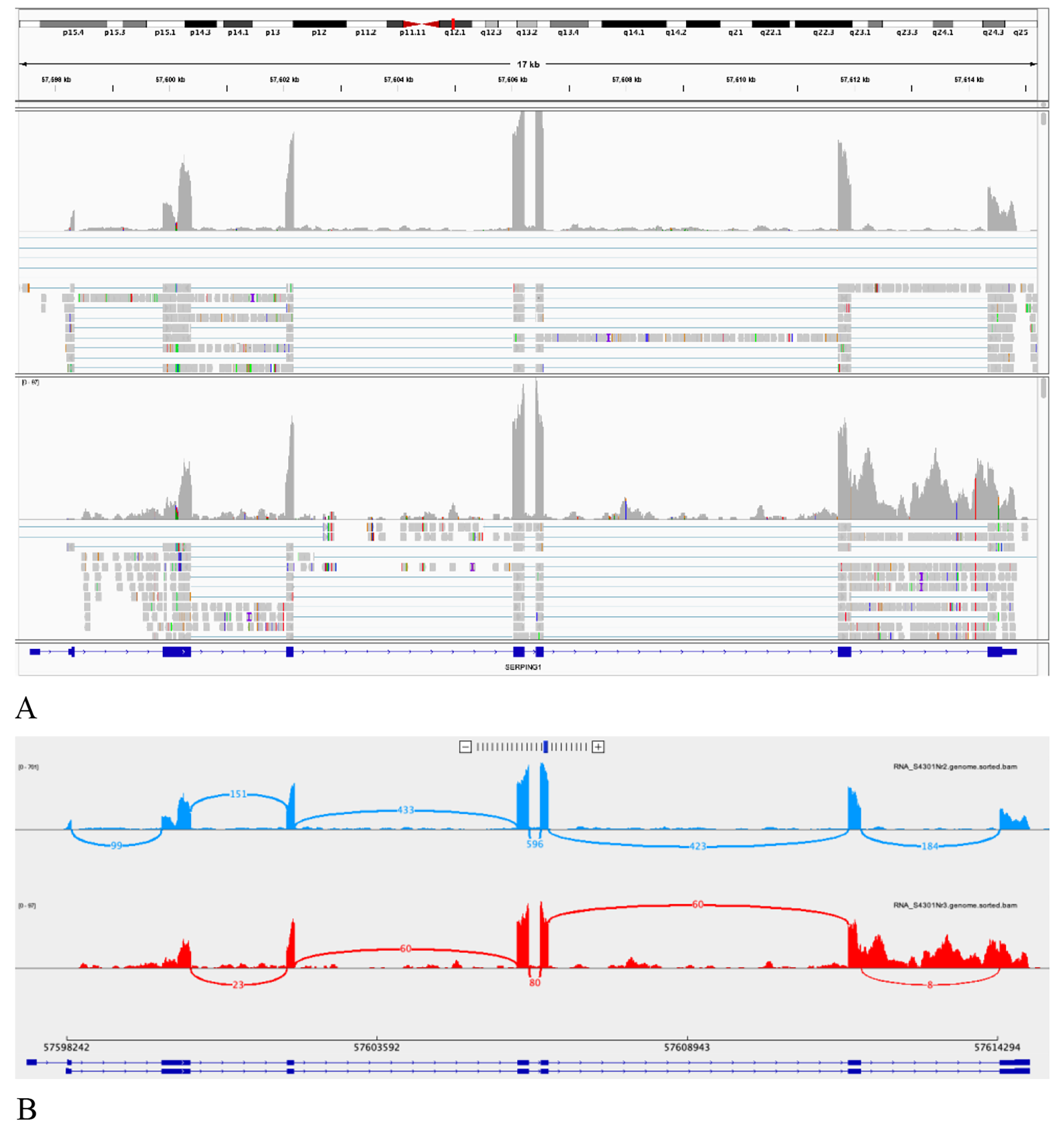
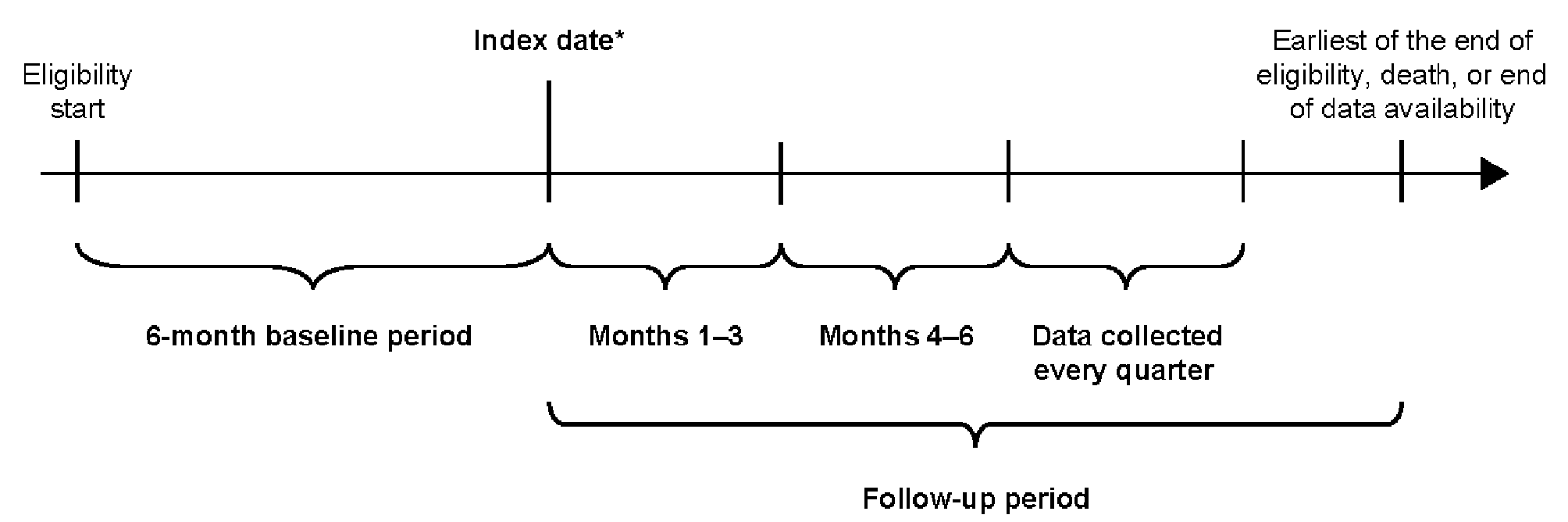
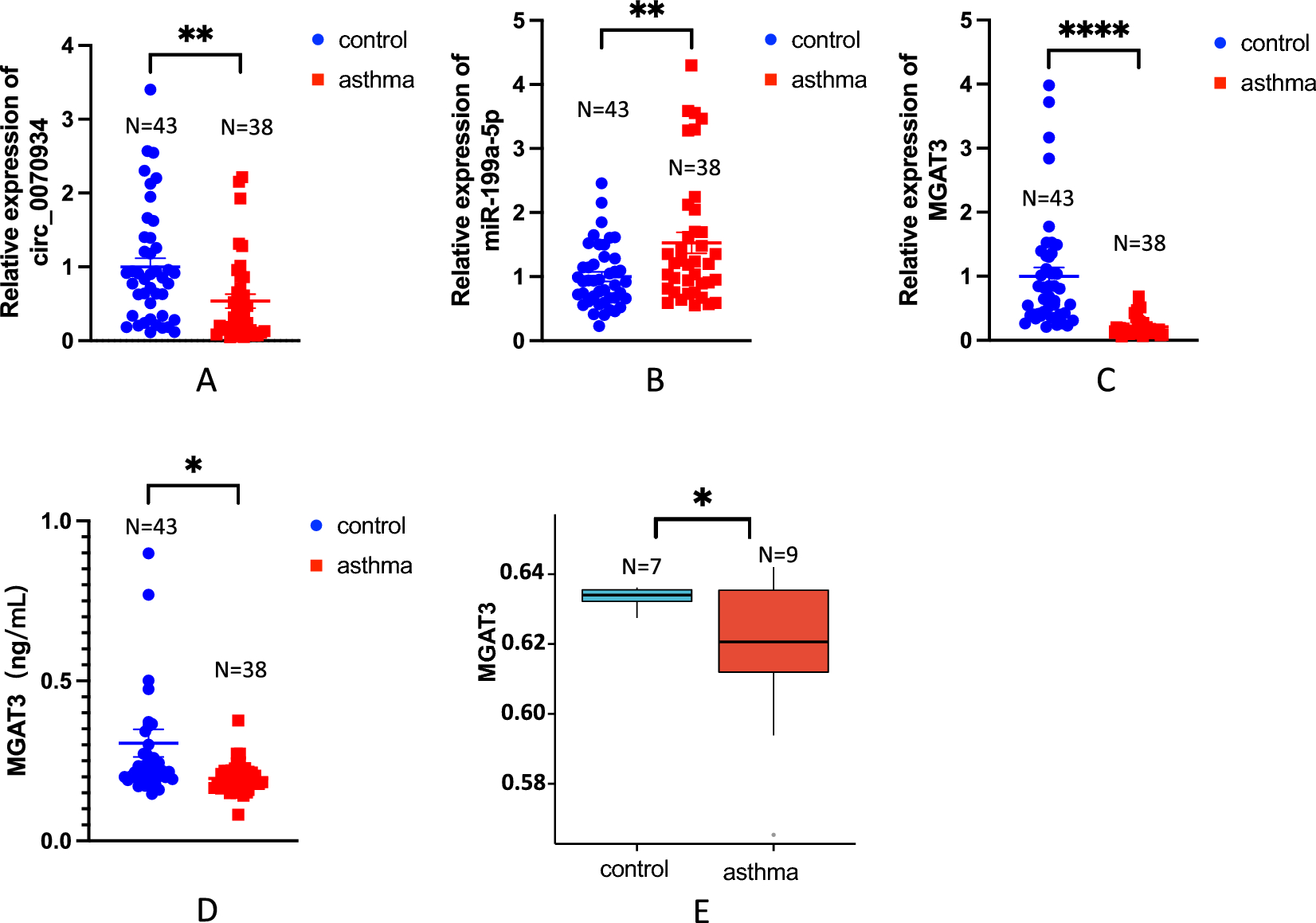
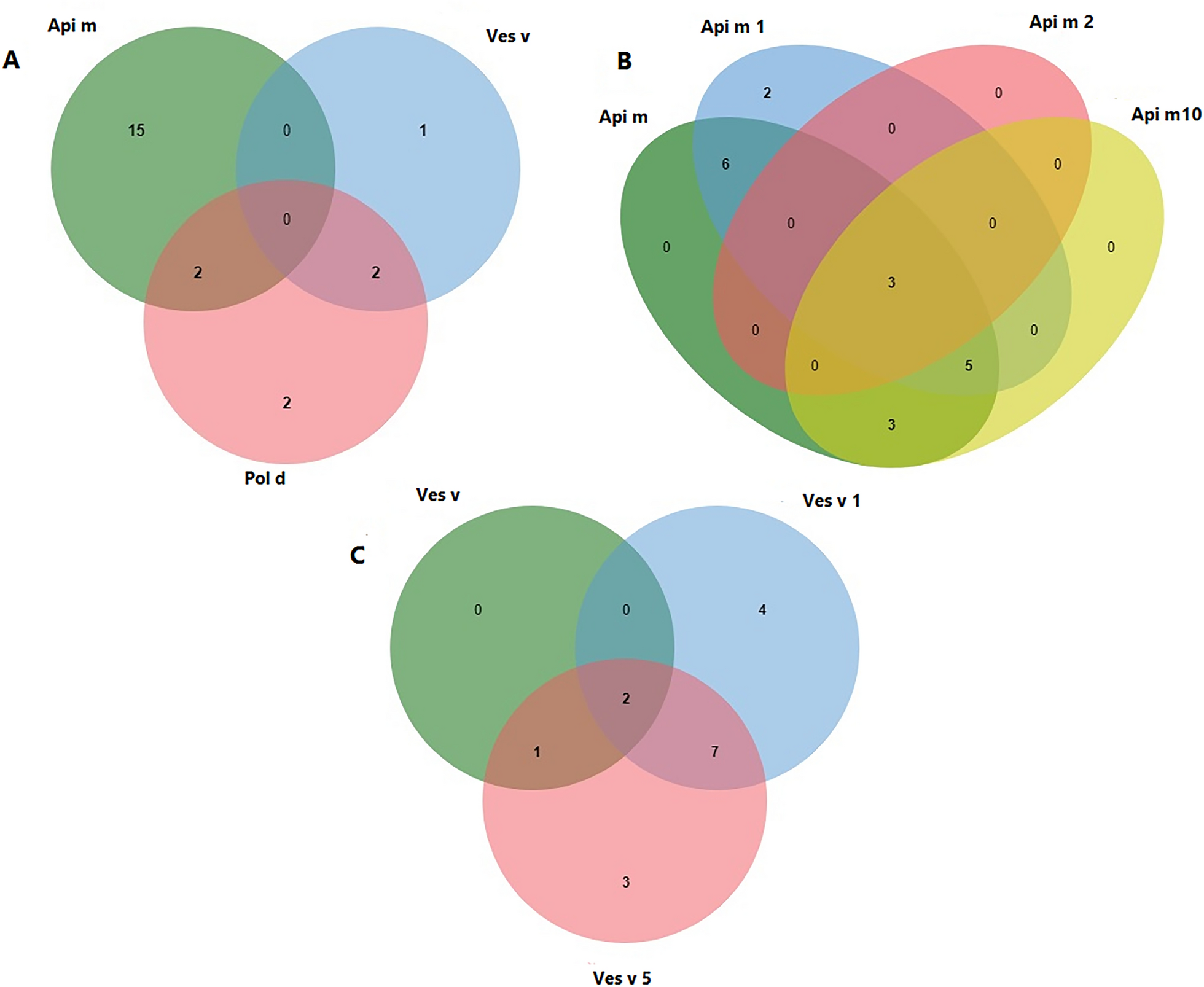
記住我

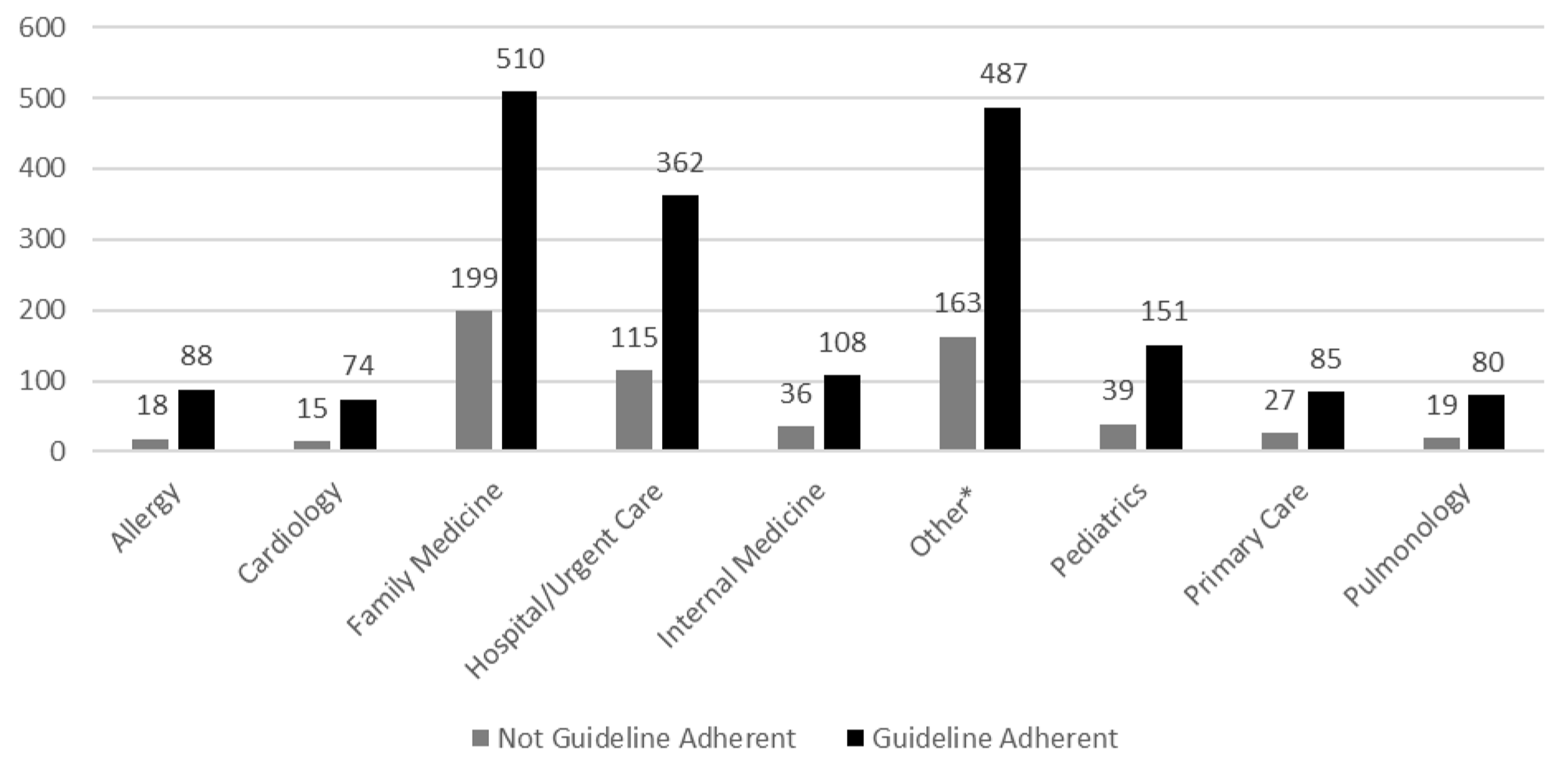
DIHS is a severe drug reaction characterized by cutaneous manifestations and multiple organ involvement. It often begins with a fever above 38 ℃, shortly followed by maculopapular rash or erythema multiforme. Typically, the skin lesion originated on the face, upper torso, or upper limbs and spreads to the whole body, occasionally accompanied by edema in eyelid, face, or neck [4, 5]. In severe cases, exfoliative dermatitis or erythroderma develop. Besides skin involvement, hepatosplenomegaly can be present and is frequently accompanied by liver and/or kidney injury during illness. There can be marked hematologic abnormalities, including leukocytosis (> 11*109 cells/L), atypical lymphocytosis (> 5%), and eosinophilia (> 1.5*109 cells/L). Serum immunoglobulin levels, including IgG, immunoglobulin A (IgA), and IgM, decreased at onset and reached their lowest levels several days after cessation of offending drugs. Subsequently, serum IgG level increases significantly after 1–2 weeks and returns to normal upon complete disease remission [6]. DIHS has a polymorphous dermatopathological appearance. A dense superficial and/or perivascular lymphocytic infiltrate is commonly present, sometimes accompanied by eosinophils, neutrophils, or extravasated erythrocytes [7]. In addition, the spongiosis or acanthosis in the epidermis was frequently consistent [8, 9]. Occasionally, a sufficiently dense lymphocytic infiltrate or the presence of atypical lymphocytes mimics lymphoma [10, 11]. Other histopathological features include interface vacuolization, lichenoid infiltrate, and individually apoptotic keratinocytes [12]. DIHS usually occurs three weeks to three months after offending drug administration, later than most other drug reactions [1]. However, symptoms may prolong and relapse even after withdrawal of offending drug [13]. Multiple organ involvement differentiates DIHS from common drug eruptions, including lymphatic, hepatic, nephritic, cardiac, neurologic, and endocrine systems [14]. Furthermore, severe hepatitis is the main cause of mortality. Due to multiple organ abnormalities, DIHS may be misdiagnosed for acute infectious diseases, lymphoproliferative diseases, and autoimmune diseases.
The absence of polyneuropathy and polyneuropathy ruled out POEMS syndrome. And, Castleman disease was unlikely due to the absence of pathologic manifestation of lymph nodes and bone marrow.
Although DIHS pathogenesis remains ambiguous, it is generally regarded as a hypersensitive reaction and sequential reactivation of HHV-6. Typical DIHS exhibits a bimodal clinical course, with the first stage including drug hypersensitivity reaction and the second stage involving reactivation of HHV-6 [1, 10]. Additionally, EBV, CMV, and HHV-7 are reactivated during the course of DIHS [15, 16]. Both Hashimoto and Shiohara groups investigated HHV-6 reactivation in severe cutaneous adverse drug reactions, detecting HHV-6 DNA only in DIHS [16, 17]. As a result, J-SCAR group established all seven diagnostic criteria for DIHS, including HHV-6 reactivation [1]. In this patient, HHV-6 DNA and IgM antibody for coxsackievirus, EBV and CMV, were not detected, possibly due to inappropriate sampling timing following virus reaction.
Several evidences have demonstrated that genetic factors are implicated with DIHS, particularly specific HLAs subtypes [18]. HLA pre-screening has been shown to reduce the risk of DIHS in selceted populations when medications such as carbamazepine, allopurinol and abacavir are employed. We retrospectively summarize recent progresses in identifying pharmacogenetic associations with DIHS in different populations (Table 3) [19]. A systematic meta-analysis of pharmacogenomics studies by Chi et al. revealed that HLA-B*27:05, HLA-B*38:02, and HLA-DRB1*08:03 alleles were linked to ATD-induced agranulocytosis, particularly in MMI [20]. Additionally, our case possessed HLA-DRB1*08:03 allele. However, large-scale studies of ATD-induced DIHS must assess the possible genetic association with HLA. 3D structural modeling was conducted to observe interactions between HLA proteins and ATD drugs [21]. In the pocket of HLA-DRB1*08:03, one asparagine residue was predicted to stabilize ATD drug molecules by bonding with sulfur and nitrogen atoms of the drug. However, the detailed mechanisms underlying ATD-HLA complex and the determinants of DIHS response remain unknown. Additional experiments are required.
Table 3 Pharmacogenetics of HLA-associated DIHSSystemic corticosteroid therapy is currently the most common treatment for DIHS. Systemic steroid therapy is often administered at a dose of 40–60 mg/d of prednisolone or equivalent. The dosage should then be tapered over 6–8 weeks to prevent relapse or organic damage [13]. When the aforementioned steroid therapy falis to relieve or exacerbates symptoms, patients may be treated with pulsed intravenous methylprednisolone (30 mg/kg for 3 days), IVIG, plasmapheresis, or a combination of the above therapies [22]. The patient, in this case, was treated with an insufficient course of steroid therapy (equivalent to intravenous prednisolone, 50 mg/d for nine days), which resulted in a repeated outbreak and protracted course of the disease. However, possibly due to a less serious condition and combined treatment with IVIG, the patient recovered. IVIG contains anti-virus IgG, which could work against HHV-6 [23]. IgG has been reported to block Fas–Fas ligand-mediated keratinocytes apoptosis [24]. Additionally, IgG has an anti-inflammatory function by increasing glucocorticoid receptor sensitivity to inhibit lymphocyte activation [25].
Until now, clinical cases of DIHS following ATD treatment have so far rarely been reported [26, 27]. We identified three cases (our case and two reported cases) on DIHS with ATDs (Table 4). Implicated drugs included MMI (n = 1) and PTU (n = 2). All three patients had a fever and diffused maculopapular rash. In the case reports, common clinical features included hematologic abnormality, liver abnormality, and lymphadenopathy. Ozaki et al. observed an increase in the titer of HHV-6 IgG and IgM antibodies, implying HHV-6 reactivation [26]. The mainstay of treatment in the three patients was corticosteroid therapy.
Table 4 Reported cases of ATDs-induced DIHSThere are two important lessons to be learned from our case. First, when treating hyperthyroidism with PTU, physicians should be vigilant for DIHS possibility, particularly late-onset drug eruption with fever, lymphadenopathy, hematological abnormalities, and multiple organ involvement. In our case, failure to promptly diagnose and initiate formal treatment resulted in symptom relapse. IgM, IgG, and DNA testing for HHV-6 has a certain clinical utility. According to condition severity, timely administration of effective treatment is critical for reducing mortality rates of DIHS. Second, the patient with the first examination exhibited Hashimoto's thyroiditis, which progressed to hyperthyroidism rather than Graves' hyperthyroidism. In general, most patients who develop Hashimoto's thyroiditis with hyperthyroidism do not require ATDs treatment. To avoid unnecessary adverse drug reactions, a cautious differential diagnosis of hyperthyroidism etiology is extremely necessary.
Fig. 1
Haematoxylin and eosin staining of the skin biopsy. Finding from the skin biopsy: dermal lymphocytic infiltrate and occasional perivascular monocytic infiltrate, with the presence of interface vacuolization (a), perifollicular lymphocytic infiltration (b), epidermal spongiosis (c). Hematoxylin and eosin stain; original magnification: X200
留言 (0)