
記住我
Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) mutations are genetic drivers in numerous cancer types including non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and pancreatic ductal adenocarcinoma (PDAC) [1,2,3,4,5]. KRAS proteins primarily bind to guanosine diphosphate (GDP) and are in an inactive conformation maintained by intrinsic guanosine triphosphate (GTP) hydrolytic activity. KRAS interacts with GTPase activating protein (GAP) accelerating GTP toward conversion of GDP [6,7,8,9], while guanine nucleotide exchange factor (GEF) binding with KRAS results in the KRAS passively loading with the GTP [8, 10, 11]. GTP binding to KRAS shifts the active site from an open to a closed conformation, allowing multiple downstream effector pathways to interact and activate, including the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways [12, 13]. The activated state of KRAS accumulating in vivo results in the activation of downstream signaling pathways and is associated with tumorigenesis, aggressive disease, and poor prognosis [14, 15].
For more than 40 years, KRAS mutation has been considered “undruggable” [16, 17]. On the one hand, the affinity of KRAS and GTP is at the pM level, while the concentration of GTP in cells is up to 0.5 μM. It is difficult to achieve effective competition like protein kinase inhibitors [18, 19]. On the other hand, the KRAS proteins are featureless. They have a nearly spherical structure that lacks a deep hydrophobic pocket and has no obvious binding site [20]. Currently, Food and Drug Administration (FDA) has approved an allele-specific covalent inhibitor of KRAS (G12C), AMG510 (sotorasib) having marked clinical responses across multiple tumor types [21,22,23,24]. In addition, a selective non-covalent inhibitor of KRAS (G12D), MRTX1133, also provides a novel targeting therapy [6, 22, 25]. In this new era of targeting KRAS mutations, the next challenge will be to understand and overcome the mechanisms of drug resistance.
In this review, we delineate the recent therapeutic strategies for KRAS mutant cancers and discuss the resistance mechanisms of KRAS mutant therapy and the possible approaches to combat them.
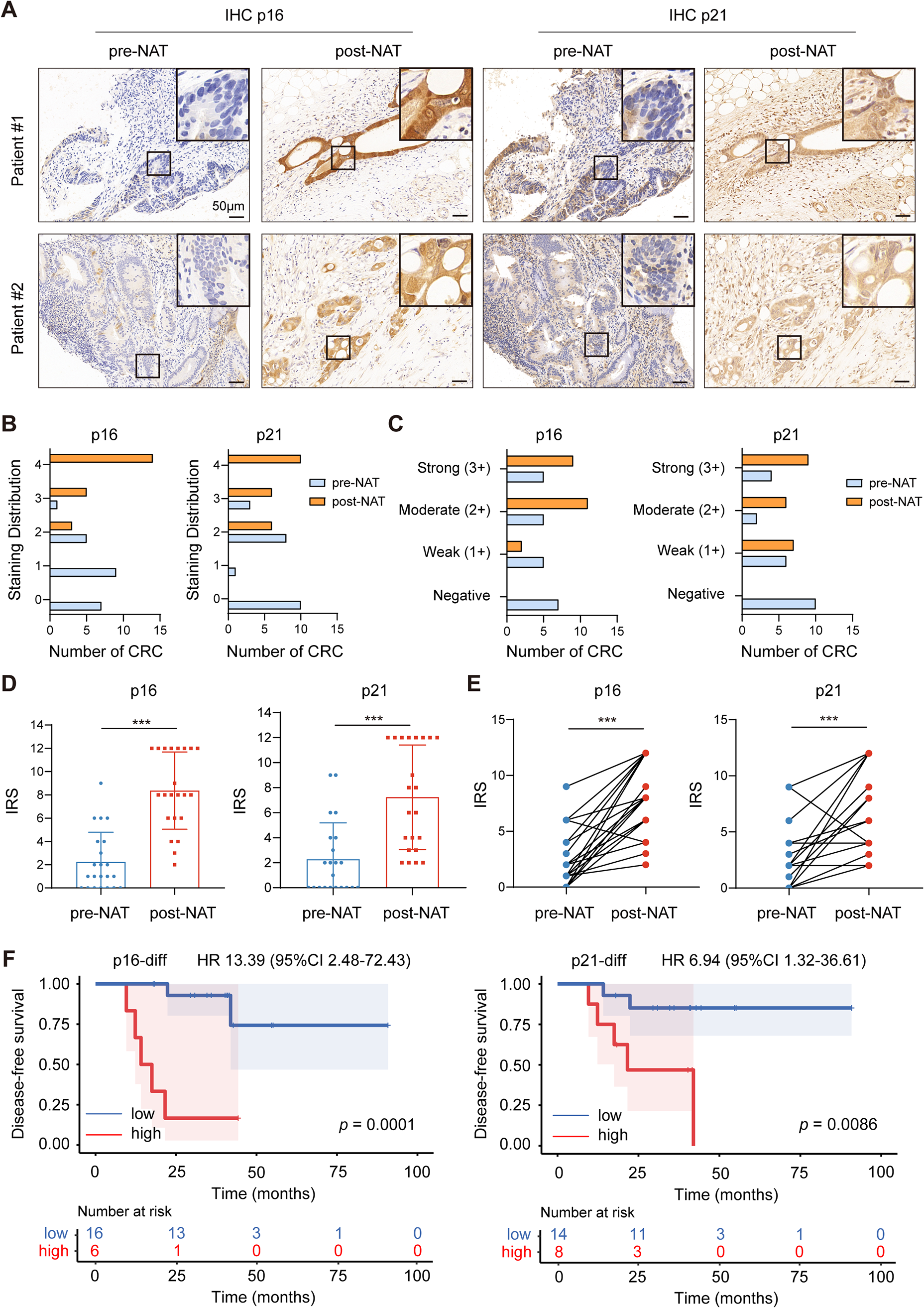
KRAS mutation-driven cancersKRAS mutations are common in a variety of cancers, for example, 45% of CRC cases in the United States and 49% of CRC cases in China; ∼90% of pancreatic ductal adenocarcinoma (PDAC) in the United States, and ∼89% in China; and 35% of lung adenocarcinomas (LUAD, a subtype of non-small-cell lung cancer) in the United States, and ∼13% in China (Fig. 1 for US data) [26]. KRAS has two isomers, KRAS4A and KRAS4B, that are generated by selective splicing of the KRAS gene. The mutant subtypes of KRAS are mainly classified as KRAS (G12D), KRAS (G12V), KRAS (G12C), KRAS (G13D), KRAS (G12R), and KRAS (G12A) mutations or KRAS wild-type amplification. Genetic alteration of G12 or G13 destroys the stability of the arginine residue hydrolysis transition state [7]. The distribution of KRAS mutations varies in different human cancers, with KRAS (G12C) mutation in 41% of LUAD, whereas KRAS (G12D) and KRAS (G12V) are the two most common alleles in CRC and PDAC, as shown in Fig. 1. Notably, other KRAS alleles such as G12R are limited in PDAC [26]. Indeed, although the tumor type is driven by KRAS mutations, its codons and the frequency of mutations vary by tissue type.
Fig. 1
Types and proportion of KRAS mutations in multiple human cancers. a Mutations of KRAS occur in different types of cancers. Data derived from the AACR GENIE 9.0 public database. b Distribution of KRAS alleles in selected tumor types. The top 6 alleles with the highest overall disease rate are listed, while the other mutations were classified in the ‘other’ category. Mutation rates for KRAS were acquired from the Cancer Facts & Figs. 2000 report published by the American Cancer Society and published articles [26]. CRC, colorectal cancer; LUAD, lung adenocarcinoma; PDAC, pancreatic ductal adenocarcinoma; IDC, invasive ductal carcinoma; STAD, stomach adenocarcinoma; UEC, undifferentiated endometrial carcinoma; EAC/GEJC, esophageal adenocarcinoma/gastroesophageal junction cancer
KRAS biology: functions and signaling pathwaysKRAS signaling provides a competitive advantage to cancer cells by participating in central carbon metabolism, increasing glucose uptake and glycolysis to increase the nutrients flux while promoting multiple branching biosynthetic pathways. It also regulates the total mitochondrial content and function by inducing phagocytosis, and the damaged mitochondrial delays tumor progression. KRAS also promotes alternative glutamine catabolism, leading to an increase in the production of nicotinamide adenine dinucleotide phosphate (NADPH) [27]. KRAS regulates pinocytosis to deal with the limited availability of mitochondrial substrates which can lead to dangerous levels of reactive oxygen species (ROS) and depleted nucleotide pools when cultured in buffered brine lacking essential nutrients. Autophagy flux provides KRAS-driven cancer cells with glutamine and glutamate to promote TCA cycling and support nucleotide production [28, 29].
Much more studies have revealed that KRAS is regarded as a switch for GDP-GTP regulation thus regulating the cytoplasmic signaling network and control various normal cellular processes. There are two splice variants of KRAS, KRAS4A and KRAS4B. In general, KRAS4B has higher expression levels. KRAS4A and KRAS4B are required for the initiation of tumor and may also have specific functions in tumor microenvironment. For example, KRAS4A expression increases tumor cell adaptation to stress, such as hypoxia. On the other hand, KRAS4B is expressed in both stem and progenitor cells. Recent studies have revisited the role of KRAS4A and KRAS4B in tumorigenesis [10, 30, 31].
As mentioned above, KRAS is a small guanosine triphosphatase (GTPase) that acts as a switch in the molecules of various cellular processes by coupling membrane growth factor receptors with intracellular signaling pathways and transcription factors. Combined with GTP, KRAS is activated, whereas KRAS is in the “off” state when binding to GDP [32]. KRAS activation is regulated by different negative and positive regulators. Negative regulators include GTPase-activating proteins (RAS-GAP), which enhances the inherent activity of KRAS-GTPase and leads to rapid hydrolysis of binding GTP [33]. Similarly, guanine nucleotide exchange factors (RAS-GEFs) are positive regulators that stimulate the release of bound GDP and exchange of GTP, resulting in the production of active KRAS-GTP complexes in response to upstream stimuli. The three main RAS-GEF families are SOS, RAS-GRF, and RAS-GRP. The SOS proteins are involved in downstream signaling transduction of receptor tyrosine kinases (RTKs), and the RAS-GRF protein is involved in Ca2+ influx/calmodulin-dependent activation of RAS and is mainly expressed in the central nervous system. The RAS-GRP activates RAS proteins downstream of non-receptor tyrosine kinases that are mainly expressed in hematopoietic cells [34].
Upstream signaling pathways of KRAS mainly include cell surface receptors, such as epidermal growth factor receptor (EGFR (ERBB1)), human epithelial growth factor receptor 2 (HER2 (ERBB2)), HER3 (ERBB3), and ERBB4. They transmit signals through KRAS after receiving external signals. This process stimulates cell proliferation and migration [35, 36].
Downstream signaling pathways mediated by KRAS mainly include rapidly accelerated fibrosarcoma (RAF) - mitogen-activated protein kinase kinase (MEK) -extracellular regulated protein kinases (ERK) and PI3K-serine/threonine-protein kinase (AKT) - mammalian target of rapamycin (mTOR) pathways. In the RAF-MEK-ERK pathway, activated KRAS-GTP rapidly increases the number of serine/threonine-specific protein kinase (RAF) from the cytoplasm to the plasma membrane, and then changes its conformation to activate it. The c-terminal catalytic domain of RAF binds to MEK1/2 and then activates ERK1/2 by phosphorylation to regulate cell proliferation, differentiation, and migration [37]. In the PI3K-AKT-mTOR pathway, KRAS-GTP binds to the p110s site of PI3K, which activates PI3K and promotes the conversion of phosphatidylinositol 4, 5-diphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 promotes AKT phosphorylation by phosphoinositide-dependent kinase 1 (PDK1) and activates mTOR, thus affecting cell proliferation, protein synthesis, survival, metabolism, transcription, and other life activities [38, 39]. In short, mutations in KRAS disrupt the guanine exchange cycle, resulting in KRAS “locking” in the active GTP-binding state, thereby activating downstream signaling pathways.
Therapeutics for KRAS mutant cancersInhibitors directly targeting KRASThe KRAS mutant proteins that drive cancer development are highly similar in sequence and structure based on the structural, mutational, and biochemical data of Harvey-RAS (HRAS). Direct inhibitors are most likely to bind to the catalytic domain of KRAS [40]. Research on direct inhibition of KRAS mutations date back to the discovery of RAS-activated mutations in human cancer cells in the 1980s when RAS-activated mutations were found in human cancer cells. However, some studies have found that KRAS mutants can be targeted by heterogeneous sites, to develop covalent inhibitors of KRAS mutants. The discovery of inhibitors that selectively target KRAS (G12C) while preserving the wild-type or other mutant KRAS is a breakthrough in the research field [20, 24].
AMG510 is a first-in-class small molecule inhibitor of KRAS (G12C) that specifically and irreversibly locks KRAS in an inactive GDP binding state, as shown in Fig. 2 [41]. It was found that the potential inhibitor of KRAS (G12C) with pocket binding histidine-residues can be flipped upward to reveal hidden grooves produced by another orientation of His95. In contrast to the ARS-1620, the first confirmed direct inhibitor of KRAS (G12C), which occupies a smaller pouch and is less potent, AMG510 enhances its binding to KRAS (G12C) through the His95 groove with an approximately 10-fold increase in potency [42] (Table 1).
Fig. 2
Structures of KRAS surfaces targeted by KRAS mutant inhibitors. a Switch-II pocket (purple) of KRAS (G12C) bound to AMG510 (PDB: 6OIM). b MRTX1133 with KRAS G12D/GDP (PDB: 7RPZ)
Table 1 Summary of KRAS mutation cancers therapeuticsMRTX849 (adagrasib) developed by Mirati Therapeutics, has been identified as a highly selective covalent inhibitor of KRAS (G12C) and is currently in phase I/II clinical studies [44, 45]. It is an oral, small-molecule selective inhibitor of KRAS (G12C) mutation. It can not only inhibit KRAS mutation almost completely in vivo but also show good drug-like properties. MRTX849 selectively targets the mutant cysteine 12 of KRAS in GDP, which is present in the induction-switch II pocket of KRAS (G12C), thereby locking it into an inactive GDP-binding state and inhibiting the RAS/MAP kinase pathway. In KRAS (G12C) positive cell lines and patient-derived xenograft models from multiple tumor types, 65% of the models showed significant tumor regression (Table 1) [60].
MRTX1133 was identified as a potent, selective, noncovalent inhibitor of KRAS (G12D) with picomolar binding affinity. Asp12 of KRAS (G12D) has a carboxyl group that is weaker nucleophilic than the sulfhydryl group of cysteine. This difference results in compounds, such as MRTX849, with significant effects on KRAS (G12C), but not on KRAS (G12D). Based on the structure of MRTX849, the electrophilic receptor, acrylamide, was replaced with piperazine to form intermolecular ion-pair force. MRTX1133 binds to the switch-II pocket and inhibits the protein−protein interactions necessary for the activation of the downstream pathway of KRAS [25] (Table 1).
Targeted regulation of KRAS active proteinThe signal transduction process of KRAS activation and inactivation is catalyzed by various factors and enzymes. The KRAS activity can be indirectly reduced by inhibiting the function of factors or enzyme activity, which achieves the purpose of inhibiting pathway activation. Son of sevenless 1 (SOS1) and SH2-containing protein tyrosine phosphatase (SHP2) are the two most critical targets in the RAS signaling pathway [61, 62].
Molecules interrupting KRAS cell membrane localization or dimerizationIt was observed that RAS is active when localized to the cell membrane. In this case, RAS requires three enzymes including isoprenylcysteine carboxyl methyltransferase (ICMT). Cysmethynil, a small molecule inhibitor of ICMT, disrupts RAS membrane binding and reduces cell growth in RAS mutant cell lines [46,47,48]. In addition, a small molecule, Cmpd2, which interferes with the binding of the RAS and lipid membranes, promotes membrane occlusion, and reduces binding to the RBD domain of RAF. What’s more, the RAS family members are oligomerized or dimerized for efficient RAS-driven signaling. Without interfering with RAS localization and GTPase activity, NS1 disrupts the self-binding of HRAS and KRAS by directly binding to the α4–α5 interface, reducing the activation of downstream pathways and inhibiting cell proliferation [30, 63, 64].
SOS1 inhibitorsIn the RAS-GEF family, SOS protein is widely expressed and participates in downstream signaling transduction of RTKs. The human SOS family contains two different genes, SOS1 and SOS2, that are located on different chromosomes [65]. Hillig et al. found an effective and cell-active small molecule inhibitor, Bay-293, which is developed by Bayer and is currently in preclinical studies [61]. Bay-293 can effectively disrupt the interaction between KRAS and its exchange factor SOS1. It interrupts the reloading of KRAS and GTP by blocking the formation of the KRAS-SOS1 complex, leading to anti-proliferative activity [61]. The results showed that Bay-293 inhibited RAS activity in Hela cells and had high anti-proliferative activity against wild-type cell lines K562 and MOLM-13 and KRAS (G12C) mutant cell lines NCL-H358 and CALU-1 [61].
Hofmann et al. reported the discovery of a highly efficient, selective, and orally bioavailable small molecule SOS1 inhibitor BI-3406, which binds to the catalytic domain of SOS1 and thus prevents its interaction with KRAS [66]. BI-3406 reduces GTP-RAS formation, thereby limiting cell proliferation in a variety of KRAS-driven cancers. Most importantly, BI-3406 also attenuates MEK-in feedback reactivation, thereby enhancing the sensitivity of KRAS-mutant cancers to MEK inhibition [66].
SHP2 inhibitorsSHP2 is the first protein tyrosine phosphatase discovered to promote the development of cancer, which is closely associated with the occurrence of breast cancer and lung cancer. SHP2, as an oncogene, is located at the downstream common node of RTKs and mediates the activation of RAS-ERK signaling pathway, thereby promoting the proliferation of cancer cells. Currently, there are four SHP2 inhibitors in clinical trials: Jabi-3068, TNO155, RC-4630, and RLY 1971. These inhibitors bind to a region outside the PTP catalytic pocket and show great selectivity against other members of the phosphatase family [67].
Chen et al. reported an efficient, selective, and orally bioavailable small molecule SHP2 inhibitor, SHP099, which controls SHP2 in the self-inhibitory structure. SHP099 binds to the interface of N-terminal SH2, C-terminal SH2, and protein tyrosine phosphatase domains simultaneously, thereby inhibiting SHP2 activity through an allosteric mechanism. SHP099 inhibits the MAPK signaling pathway that results in inhibiting tyrosine kinase receptor-driven proliferation of human cancer cells in vitro and a mouse tumor transplant model in vivo [68].
The SHP2 inhibitor developed by Novartis, TNO155, inhibits MAPK signaling and enhances the efficacy of KRAS (G12C) inhibitors against KRAS (G12C) lung and colorectal cancers. The tumor microenvironment can be affected by blocking immunosuppressive signals RTKs and MAPK signals, thus reducing the overexpression of SHP2 and slowing down tumor growth [49].
Inhibitors of KRAS upstream signaling pathwayEGFR as an RTK plays a vital role in cell proliferation and migration. Most of the signaling transduction of EGFR is thought to occur in the plasma membrane, stimulating the activation and signaling transduction of MAPK and PI3K. Ligand-mediated EGFR activation is through conformational changes in the extracellular domain of the receptor following ligand binding, leading to receptor dimerization and the formation of kinase domains that internalize asymmetric dimers, while EGFR without ligand can also internalize, but at a relatively slow rate. Two classes of EGFR-targeting compounds, monoclonal antibody (mAb) like cetuximab, and tyrosine kinase inhibitor (TKIs) like gefitinib targeting the extracellular and intracellular domains of EGFR, have shown antitumor activities [69].
Gefitinib, reported by Mohamed Muhsin, is a first-generation small molecule EGFR inhibitor that binds to the intracellular tyrosine kinase domain of EGFR, thereby inhibiting autophosphorylation of receptor and then isolating the downstream signaling transmission [58]. Afatinib has clinical activity as a second-generation inhibitor against major uncommon and complex EGFR mutations in NSCLC [70].
AZD9291, reported by Cross et al., is a novel oral, potent, and selective third-generation irreversible inhibitor that inhibits EGFR (T790M) resistant mutation without affecting wild-type EGFR [71]. The Janne’s clinical trial of AZD9291 involved 253 patients. In 31 patients enrolled in the dose-escalation cohort, no dose-limiting toxic effects happened under the assessed dose [72]. An additional 222 patients were treated in five extended cohorts. The overall objective tumor response rate was 51%, indicating that AZD9291 was highly effective in patients with EGFR (T790M) mutant lung cancer whose disease had progressed during previous treatment with EGFR tyrosine kinase inhibitors [71, 72]. JBJ-04-125-02, as an EGFR-mutant allosteric inhibitor, inhibits EGFR (L858R/T790M/C797S) pathway and cell proliferation. However, increased dimeric EGFR reduces the efficacy, leading to drug resistance [73].
Inhibitors of KRAS downstream signaling pathways RAF-MEK-ERKExcept downstream of post-translational KRAS and membrane-binding processes, other key targets are KRAS protein mutation-activated signaling pathways. One of these pathways is the RAF-MEK-ERK pathway, and several MEK inhibitors have been developed [74]. For example, trametinib (GSK112021) is a selective alloy structural oral inhibitor that inhibits MEK1/2 activation and kinase activity. Selumetinib, another oral MEK1/2 inhibitor, had no significant effect on survival compared to capecitabine in gemcitabine refractory PDAC patients. Lifafenib (BGB283) is a novel experimental inhibitor of RAF dimer with effective and reversible inhibition of the wild-type A-RAF, B-RAF, C-RAF, B-RAFV600E as well as EGFR and KRAS. A dose-escalation/dose-expansion study in humans by Desai et al. evaluated the role of sorafenib in solid tumors with B-RAF and KRAS mutations. Antitumor activity was shown in KRAS-mutated NSCLC (response rate 16.7%) and endometrial/ovarian cancer (DCR 100%) [75].
Diamond et al. validated Cobimetinib, an oral inhibitor of MEK1 and MEK2 in a clinical trial. Among the 18 patients treated, the overall response rate was 89% and there was no acquired drug resistance. After 1 year, 94% of patients maintained the characteristics of progression-free tumors, demonstrating the efficacy of Cobimetinb in treating tumors by acting on the MAPK pathway [68].
Smetinib, another oral inhibitor of MEK1/2 developed by AstraZeneca, reduced the size of plexus neurofibroma (PNF) by at least 20% from baseline in 71% patients in the Phase 2 trial (SPRINT). In a Phase 2 trial with low-grade glioma (LGG), partial remission of PNF was found in 40% of patients and 2-year progression-free survival achieved in 96% of patients. In gemcitabine refractory PDAC patients, Selumetinib had no significant effect on survival compared with Cobimetinb [76, 77].
Ulititinib (BFD − 523) is an ERK1/2 kinase inhibitor with strong preclinical activity in cell lines with BRAF and RAS mutations. In phase I clinical trials, it has an acceptable safety profile and good pharmacokinetics, and is active against solid tumors with NRAS and BRAFV600 and non-V600 mutated cancers [78].
PI3K-AKT-mTORAnother effector, PI3K, is also activated by KRAS. Unlike p110γ and p110δ, p110α is widely expressed and exclusively activated by RAS. As isoform-specific p110 inhibitors should target malignant cells more specifically, they are expected to have fewer off-target effects. Alpelisib (BYL719) is a p110α-specific inhibitor tested in a phase I trial including patients with PIK3CA-altered advanced solid tumors [10, 79].
MK2206, an AKT inhibitor, was found to improve the pathological complete remission (CR) rate in patients with breast cancer associated with positive hormone receptors [
留言 (0)