
記住我
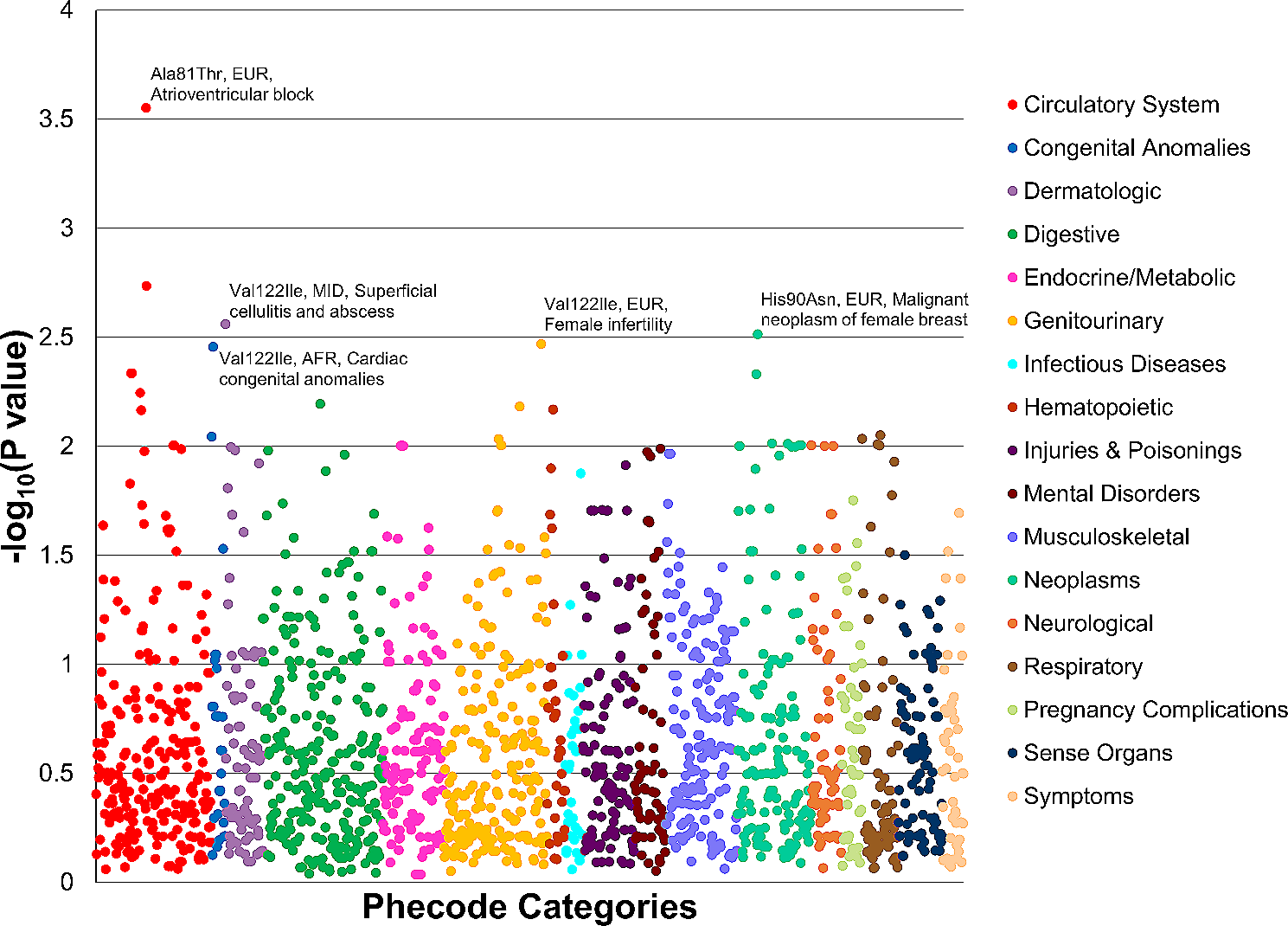
The IndiGenomes dataset comprises about 56 million genetic variants from the genomes of 1029 self-declared healthy Indian individuals. Out of these, over 18 million genetic variants are unique to the Indian population [21]. We used this dataset and extracted a total of 186,782 variants present in 36 cardiac ion channelopathy-associated genes (Additional file 1).
For analysis of nonsynonymous variations, ANNOVAR annotations on the IndiGenomes dataset were used [21]. By applying a variant filtering pipeline (Fig. 1), we analysed the spectrum of rare and probably pathogenic variants associated with cardiac ion channelopathies. Firstly, by applying a conservative cut-off of MAF < 0.05 in major population datasets, we obtained a compendium of 156,351 rare variants. Out of these, 1263 were exonic variants. Their mapping with respect to the reference gene annotations has been summarised in Additional file 3. Next, we retrieved all the exonic nonsynonymous SNVs which accounted for a total of 693 variants. To obtain the probably pathogenic variants, we selected predicted deleterious variants as annotated from SIFT, PolyPhen or CADD and removed benign or likely benign variants reported in the ClinVar database. As a result, we were left with a corpus of 440 nonsynonymous variants.
Interpretation of nonsynonymous variants according to ACMG/AMP guidelinesA total of 440 nonsynonymous exonic variants in 36 genes were obtained after applying the variant filtering pipeline. These were taken ahead for in-depth genetic interpretation based on the ACMG/AMP guidelines [32] and the final classification was carried out using Genetic Variant Interpretation Tool [33].
A subset of 36 variants was classified as pathogenic (n = 1) or likely pathogenic (n = 35). Depending on the available strength of evidence, particularly in the context of functional studies, the variants in the likely pathogenic category were divided into high confidence (likely pathogenic (II/III), n = 6) and low confidence (likely pathogenic (IV/V), n = 29) (Fig. 2A). Moreover, 16 variations were classified as likely benign, out of which, 13 variations were present in KCNH2 (n = 6) and PKP2 (n = 7) genes. A majority of variants (n = 388) were classified as variants of uncertain significance (VUS). This category was assigned in cases where there was a lack of evidence or conflicting evidence for interpreting the pathogenicity of the variant. Of all the variants classified as VUS, more than half (58.2%) were harboured in 6 genes: AKAP9, ANK2, RYR2, SCN10A, SCN5A and TRPM4 (Fig. 2C).
Fig. 2
Distribution of ACMG/AMP classified variants associated with cardiac ion channelopathy. A Pie chart representing the percentage of nonsynonymous variants classified as VUS, LP(IV) or (V), P/LP(II) or (III) and LB. B Pie chart representing the percentage of pLoF variants classified as VUS and P. C Heat map representing the distribution of 470 ACMG/AMP classified variants (nonsynonymous and pLoF) across 36 cardiac ion channelopathy genes. Red colour gradient corresponds to the number of variants in a gene. Numbers in rectangles represent the number of variants
In total, we have identified 7 pathogenic or high confidence likely pathogenic nonsynonymous variants. These variants were found to be present in 12 out of the 1029 individuals analysed. The variants KCNH2:p.R823W, SCN5A:p.V2016M and CACNA1C:p.R858H were present in 1 individual each whereas the variants KCNE2:p.I57T, KCND3:p.L450F and KCNE3:p.V17M were present in 2 individuals each. The variant SCN3B:p.V110I was present in 3 individuals. All of the 12 individuals carry the respective variants in heterozygous state. Additionally, all these 7 variants have been independently validated by sanger capillary sequencing (primer details mentioned in Additional file 4). Considering that the disorders associated with the above-mentioned genes follow an autosomal dominant mode of inheritance, individuals carrying the variants in these genes may be at risk of developing a channelopathy disorder.
The details of all 36 P/LP variants are summarised in Table 1. The complete list of 440 nonsynonymous variations with their ACMG/AMP annotations is elaborated in Additional file 5.
Table 1 Pathogenic and likely pathogenic cardiac channelopathy variants as per ACMG/AMP guidelinesOwing to incomplete penetrance of the disease, strong evidence to ascertain pathogenicity in the context of segregation studies was lacking in the literature for almost all of the variations. Thus, functional studies became the major determinant. For instance, a p.R823W variation in the KCNH2 gene was classified as a pathogenic variant as it has been demonstrated to cause trafficking defects by in vitro study [36] and loss of function phenotype using zebrafish [37] as well as high-throughput electrophysiological phenotyping [38]. Another variation, p.V2016M in the SCN5A gene was classified as high confidence likely pathogenic since it had been shown to reduce cell surface expression and peak Na + currents in HEK293 cells [39]. Moreover, mice experiments in the same study have shown that the SIV domain spanning the amino acid valine plays an important role in the correct expression of Nav1.5 in the lateral myocyte membrane, which is further important for cardiac conduction. Another study for the p.V2016M variation reported that it exhibits loss of function as well as gain of function features by protein kinase A activation or C activation [40].
Interpretation of predicted loss of function variantsLoss of function variants include splicing, stopgain and frameshift variants. They can have deleterious effects on the protein function and thus, can potentially cause the disease. We evaluated pLoF variants separately in the IndiGenomes dataset which mapped to the 36 cardiac ion channelopathy genes. Variants were annotated from Variant effect Predictor tool (VEP) and using loss of function transcript effect estimator (LOFTEE), we predicted 30 high confidence LoF variants in the canonical transcripts of the respective genes. These variants were present in 14 genes and the list included 10 splice-site, 11 stopgain and 9 frameshift variants. Systematic annotation according to the ACMG/AMP guidelines yielded 6 variants as pathogenic and the remaining 24 variants as VUS (Fig. 2B).
Pathogenic variants were revealed in CASQ2 (n = 2), KCNQ1 (n = 2), TRDN (n = 1) and PKP2 (n = 1) genes. Of the two variants found in the CASQ2 gene, one was a stopgain variant, p.E236* and the other was a splicing variant c.420 + 2 T > C. Calsequestrin (CASQ2) is a calcium binding protein in the sarcoplasmic reticulum of cardiomyocytes and plays a key role in calcium homeostasis. We identified two pathogenic frameshift variations, p.W120* and p.G179Sfs*62 in the KCNQ1 gene. Loss of function variations in this potassium channel encoding gene are associated with the disease phenotype. Furthermore, stopgain variants p.Q513* and p.R413* were noted in TRDN and PKP2 genes, respectively. The Triadin (TRDN) is an important component of the calcium release unit in the sarcoplasmic reticulum of cardiomyocytes that interact with both ryanodine receptor (RYR2) as well as calsequestrin (CASQ2). Plakophilin2 (encoded by PKP2) is a desmosomal protein found in the intercalated discs of cardiac cells. The p.R413* variation was first identified in a Caucasian male with arrhythmogenic right ventricular cardiomyopathy (ARVC) [41]. Later on, Alcalde et al. in 2014 reported the same variant to be segregating in a Hispanic family with ARVC [42]. The summary of pathogenic pLoF variants is outlined in Table 2. All of these pathogenic variations were predicted to cause loss of protein function and deleterious by the CADD tool. The corresponding genes have an established loss of function mechanism for causing the disease.
Table 2 Pathogenic pLoF cardiac channelopathy variants as per ACMG/AMP guidelinesThe 6 pathogenic cardiac channelopathy-associated pLoF variants were found to be present in 7 out of 1029 individuals. The variants CASQ2:p.E236*, CASQ2:c.420 + 2 T > C, PKP2:p.R413*, KCNQ1:p.W120* and KCNQ1:p.G179Sfs*62 were present in 1 individual each whereas the variant TRDN:p.Q513* was present in 2 individuals. All these individuals carry the respective variants in heterozygous state and the variants have been validated by sanger capillary sequencing. The primer details are summarised in Additional file 4. The variations in genes CASQ2 and TRDN are reported to be highly penetrant and follow an autosomal recessive mode of inheritance. On the other hand, variations in genes PKP2 and KCNQ1 majorly follow autosomal dominant mode. Individuals with pLoF variations in these genes may be at risk of developing the respective channelopathy disorders.
The complete details of 30 pLoF variants along with their ACMG/AMP annotations have been provided in Additional file 6.
Variants unique to the Indian population in cardiac channelopathy-associated genesInitially, we had obtained a total of 186,782 variants in 36 genes. After final filtering, the number of nonsynonymous variants reduced to 440, out of which 114 (25.9%) were unique to the IndiGenomes dataset and absent in global population datasets, publicly available databases and literature. Of these 114 unique variants, 98 (85.9%) were classified as VUS, 12 (10.5%) as low confidence, likely pathogenic, and 4 (3.5%) as likely benign. Across genes, the unique likely pathogenic variants were found in RYR2 (n = 5), KCNJ2 (n = 2), CAV3, KCND3, KCNQ1, ABCC9, and KCNE2 (n = 1 each) genes. The 4 likely benign variants were found in KCNH2 (n = 1) and PKP2 (n = 3) genes. The complete list of nonsynonymous unique variants is mentioned in Additional file 7. Similarly, in the case of pLoF variants, we observed that 10 out of 30 variants were unique to the IndiGenomes dataset. This includes 8 VUS and 2 pathogenic variants. The pathogenic variants included CASQ2:c.420 + 2 T > C and KCNQ1:p.W120*. In total, we have discovered 124 out of 470 (26.3%) variants as unique variants in the Indian population (Fig. 3). All of these variants are yet to be identified in the channelopathy patients and functionally characterised.
Fig. 3
Distribution of 124 unique variations across genes with their ACMG/AMP classification. The number of variations corresponding to the genes are plotted as distinct bars. The colours in stacks correspond to the respective classification according to ACMG/AMP guidelines
The above observations highlight that there are a significant number of variants that are unique and are represented only in the Indian population compared to the rest of the world.
Allele frequency comparison of P/LP variants across various population genome datasetsWe sought to understand the significant allele frequency differences between the Indian population dataset and rest of the global population datasets. Allele frequencies of 13 ACMG/AMP classified pathogenic and high confidence likely pathogenic variants were fetched from the IndiGenomes dataset. Out of these 13 variants, 7 were nonsynonymous (P = 1, LP = 6) and 6 were predicted loss of function (P = 6).
Three variations namely, CACNA1C:p.R858H, CASQ2:c.420 + 2 T > C and KCNQ1:p.W120* were represented only in the IndiGenomes dataset and absent in other global population datasets (Fig. 4). The remaining 10 variations were represented in the gnomad_exome_All dataset. Except for KCNE2:p.I57T and SCN5A:p.V2016M, all of them were enriched in the IndiGenomes dataset as compared to the gnomad_exome_All dataset (p value < 0.05, Fisher’s exact test). However, on comparing the IndiGenomes frequencies specifically with the gnomad_exome_SAS dataset, differences between allele frequencies were not significant suggesting that amongst the available global datasets, SAS dataset in gnomAD is a better representative of allele frequencies in Indian population.
Fig. 4
Comparison of allele frequencies of pathogenic and likely pathogenic cardiac channelopathy variants across different genomic datasets. The variant allele frequencies in different populations are plotted as solid bubbles (with filled colours). The circles outside the bubbles represent the significantly different allele frequency values (Fisher’s exact test; p < 0.05) using IndiGenomes dataset as a reference. Red asterisk: Fisher’s exact test was not done in case of ESP6500siv2_All dataset due to unavailability of allele numbers and allele counts
Only two of the variations, i.e. KCNE2:p.I57T and SCN3B:p.V110I, were represented in the 1000 genome_All dataset. In both of the cases, there were no significant differences between the allele frequencies when compared with the IndiGenomes dataset. None of the variations was present in the 1000genome_SAS dataset.
In comparison with region-specific genomic datasets such as Qatar and GME, we found that the variations KCND3:p.L450F and KCNE2:p.I57T were enriched in the Qatar and GME datasets as compared to the IndiGenomes dataset indicating a higher genotypic prevalence of Brugada syndrome-associated risk alleles in the Middle East. Lastly, only three variations, KCND3:p.L450F, KCNE2:p.I57T and SCN3B:p.V110I, were represented in the GenomeAsia100k dataset. Allele frequencies for all of them did not differ significantly as compared to the IndiGenomes dataset. The allele frequencies of P/LP cardiac channelopathy variants across different genomic datasets are provided in Additional file 8.
Intersecting pathogenic and high confidence likely pathogenic variants in an independent cardiac ion channelopathy patient cohortWe intersected the 13 ACMG/AMP classified P/LP variations obtained from the IndiGenomes dataset with the exome sequencing dataset from a patient cohort provisionally diagnosed with cardiac channelopathy disorder (n = 53). Consequently, we found that 3 out of the 13 variations were present in the patient cohort data.
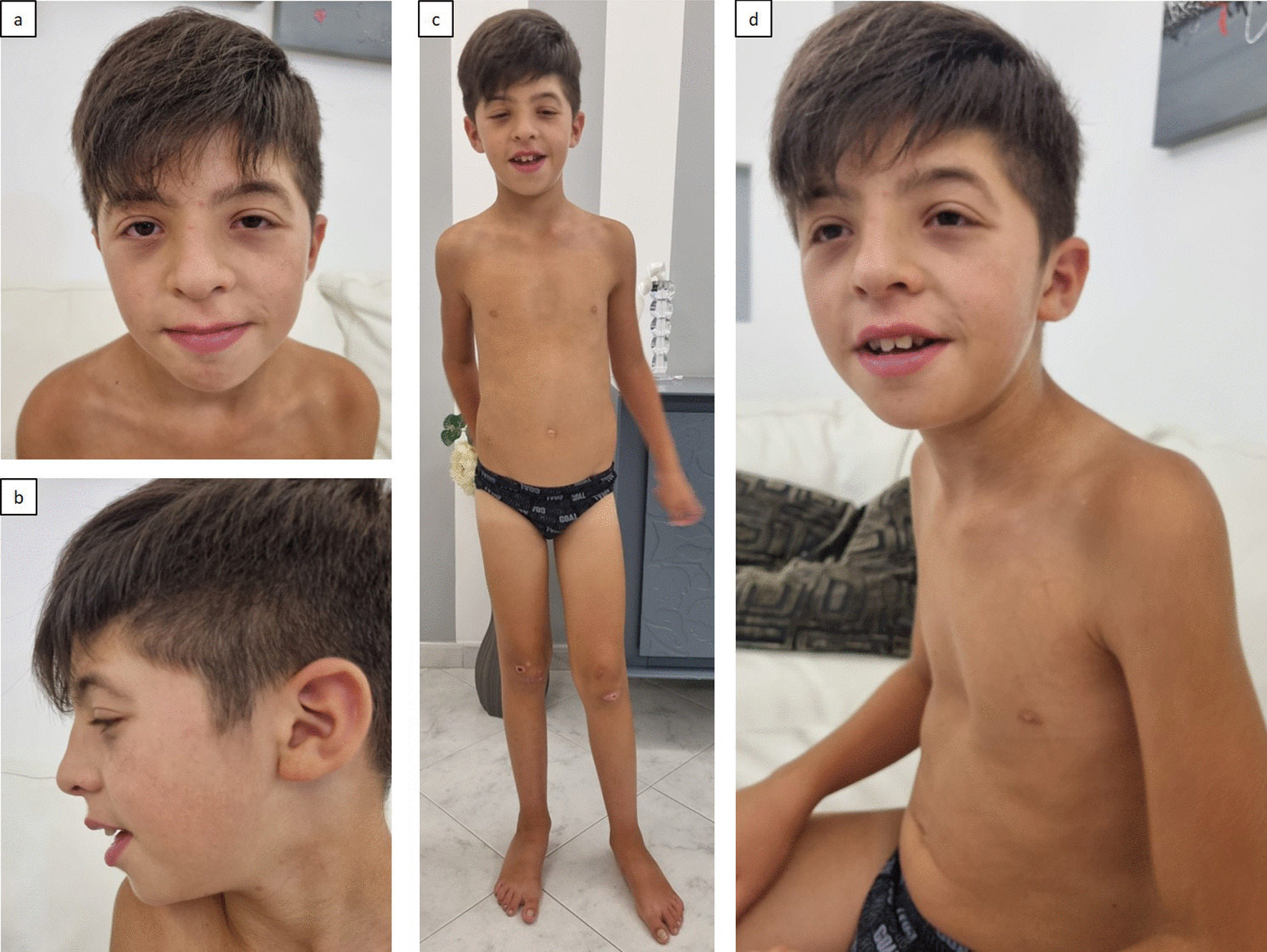
Our analysis revealed a heterozygous individual for a pathogenic frameshift variation, KCNQ1:p.G179Sfs*62. The same variation was also found in a patient with provisional diagnosis of Jervell and Lange-Nielsen Syndrome in the cardiac channelopathy cohort. The variation was present in a homozygous state in the patient. Further investigation in the cardiac channelopathy cohort revealed an LQTS patient with ECG abnormality carrying a variation, KCNH2:p.R823W which is also identified in the IndiGenomes. In addition to these two variations, a pathogenic splicing variation, CASQ2:c.420 + 2 T > C was found to be overlapping between IndiGenomes and the cardiac channelopathy cohort. The variation was found to be present in a heterozygous state in both datasets. However, the CASQ2:c.420 + 2 T > C variation was not able to explain the complete phenotypic spectrum of the patient, which is being investigated. These findings underscore the clinical utility of our analysis of channelopathy variants in the healthy Indian population.
留言 (0)