van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372(9646):1342–53.
PubMed
Article
CAS
Google Scholar
Engel AG. Acid maltase deficiency of adult life. Trans Am Neurol Assoc. 1969;94(250–2).
Engel AG. Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain. 1970;93(3):599–616.
CAS
PubMed
Article
Google Scholar
Nigro V, Aurino S, Piluso G. Limb girdle muscular dystrophies: update on genetic diagnosis and therapeutic approaches. Curr Opin Neurol. 2011;24(5):429–36.
CAS
PubMed
Article
Google Scholar
• Straub V, Murphy A and Udd B. 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord. 2018;28(8):702–10. An importance paper that redefines the classification of limb-girdle muscular dystrophies. A number of previously classified limb-girdle muscular dystrophies (LGMD) were removed from the classification because the muscle biopsies were not dystrophic or there were only small number of families defined previously. Pompe disease, that was previously classified as LGMD was removed.
Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6:177.
PubMed
PubMed Central
Article
CAS
Google Scholar
Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713–6.
CAS
PubMed
Article
Google Scholar
Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998;79(1):69–72.
CAS
PubMed
Article
Google Scholar
• Tang H, Feuchtbaum L, Sciortino S, Matteson J, Mathur D, Bishop T, et al. The first year experience of newborn screening for Pompe disease in California. Int J Neonatal Screen. 2020;6(1):9. The experience from California, the largest state in the United States, about newborn screening for Pompe disease, with appreciably higher incidence of Pompe disease and pseudodeficiency.
Burton BK, Charrow J, Hoganson GE, Fleischer J, Grange DK, Braddock SR, et al. Newborn screening for Pompe disease in Illinois: experience with 684,290 infants. Int J Neonatal Screen. 2020;6(1):4.
PubMed
PubMed Central
Article
Google Scholar
Ficicioglu C, Ahrens-Nicklas RC, Barch J, Cuddapah SR, DiBoscio BS, DiPerna JC, et al. Newborn screening for Pompe disease: Pennsylvania experience. Int J Neonatal Screen. 2020;6(4):89.
PubMed Central
Article
Google Scholar
Klug TL, Swartz LB, Washburn J, Brannen C, Kiesling JL. Lessons learned from Pompe disease newborn screening and follow-up. Int J Neonatal Screen. 2020;6(1):11.
PubMed
PubMed Central
Article
Google Scholar
Sawada T, Kido J, Sugawara K, Momosaki K, Yoshida S, Kojima-Ishii K, et al. Current status of newborn screening for Pompe disease in Japan. Orphanet J Rare Dis. 2021;16(1):516.
PubMed
PubMed Central
Article
Google Scholar
• Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ and Hwu WL. Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr. 2011;158(6):1023–27.e1. This paper details the efforts made by investigators in Taiwan to introduce NBS for a number of lysosomal disorders, including Pompe Disease. Taiwan was the first country in the world to introduce NBS for Pompe Disease and had to contend with issues with false positives from pseudodeficiency, that has a much higher prevalence in South East Asian populations.
Yang CF, Liu HC, Hsu TR, Tsai FC, Chiang SF, Chiang CC, et al. A large-scale nationwide newborn screening program for Pompe disease in Taiwan: towards effective diagnosis and treatment. Am J Med Genet A. 2014;164a(1):54–61.
Deenen JC, Horlings CG, Verschuuren JJ, Verbeek AL, van Engelen BG. The epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73–85.
PubMed
Article
Google Scholar
Johnson NE. Myotonic Muscular Dystrophies. Continuum (Minneap Minn). 2019;25(6):1682–95.
Google Scholar
Hagemans ML, Winkel LP, Van Doorn PA, Hop WJ, Loonen MC, Reuser AJ, et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain. 2005;128(Pt 3):671–7.
CAS
PubMed
Article
Google Scholar
Herzog A, Hartung R, Reuser AJ, Hermanns P, Runz H, Karabul N, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J Rare Dis. 2012;7:35.
PubMed
PubMed Central
Article
Google Scholar
•• Wencel M, Shaibani A, Goyal NA, Dimachkie MM, Trivedi J, Johnson NE, et al. Investigating Pompe Prevalence in Neuromuscular Medicine Academic Practices (The IPaNeMA Study) Neurology: Genetics. 2021;7:e623. A well-designed investigator-initiated study that assessed the prevalence of Pompe Disease in a consortium of academic/tertiary neuromuscular neurology practices. It found the prevalence of LOPD to be 1% but another 1% of patients were found to have pseudodeficiency.
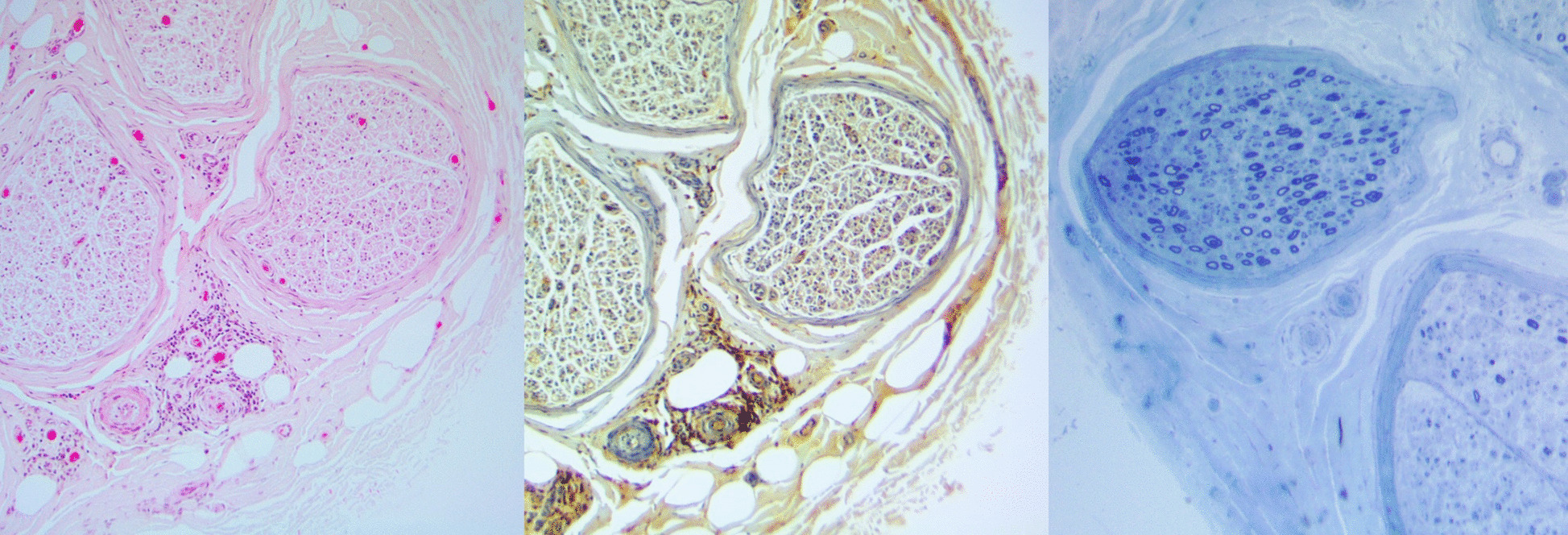
Werneck LC, Lorenzoni PJ, Kay CS, Scola RH. Muscle biopsy in Pompe disease. Arq Neuropsiquiatr. 2013;71(5):284–9.
PubMed
Article
Google Scholar
• Niño MY, Wijgerde M, de Faria DOS, Hoogeveen-Westerveld M, Bergsma AJ, Broeders M, et al. Enzymatic diagnosis of Pompe disease: lessons from 28 years of experience. Eur J Hum Genet. 2021;29(3):434–46. A nice paper describing the collective experiences from diagnosis of Pompe disease using enzymatic assays.
Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJ and Wokke JH. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology. 1999;52(4):851–3.
Kronn DF, Day-Salvatore D, Hwu WL, Jones SA, Nakamura K, Okuyama T, et al. Management of confirmed newborn-screened patients with Pompe disease across the disease spectrum. Pediatrics. 2017;140(Suppl 1):S24–45.
PubMed
Article
Google Scholar
Buehler T, Bally L, Dokumaci AS, Stettler C, Boesch C. Methodological and physiological test-retest reliability of (13) C-MRS glycogen measurements in liver and in skeletal muscle of patients with type 1 diabetes and matched healthy controls. NMR Biomed. 2016;29(6):796–805.
CAS
PubMed
Article
Google Scholar
Heinicke K, Dimitrov IE, Romain N, Cheshkov S, Ren J, Malloy CR, et al. Reproducibility and absolute quantification of muscle glycogen in patients with glycogen storage disease by 13C NMR spectroscopy at 7 Tesla. PLoS ONE. 2014;9(10): e108706.
PubMed
PubMed Central
Article
CAS
Google Scholar
Wary C, Laforêt P, Eymard B, Fardeau M, Leroy-Willig A, Bassez G, et al. Evaluation of muscle glycogen content by 13C NMR spectroscopy in adult-onset acid maltase deficiency. Neuromuscul Disord. 2003;13(7–8):545–53.
PubMed
Article
Google Scholar
Labrousse P, Chien YH, Pomponio RJ, Keutzer J, Lee NC, Akmaev VR, et al. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol Genet Metab. 2010;99(4):379–83.
CAS
PubMed
Article
Google Scholar
Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5(2):140–7.
PubMed
Article
Google Scholar
Forsha D, Li JS, Smith PB, van der Ploeg AT, Kishnani P, Pasquali SK. Cardiovascular abnormalities in late-onset Pompe disease and response to enzyme replacement therapy. Genet Med. 2011;13(7):625–31.
CAS
PubMed
PubMed Central
Article
Google Scholar
Montagnese F, Granata F, Musumeci O, Rodolico C, Mondello S, Barca E, et al. Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J Inherit Metab Dis. 2016;39(3):391–8.
PubMed
Article
Google Scholar
Pichiecchio A, Sacco S, De Filippi P, Caverzasi E, Ravaglia S, Bastianello S, et al. Late-onset Pompe disease: a genetic-radiological correlation on cerebral vascular anomalies. J Neurol. 2017;264(10):2110–8.
CAS
PubMed
Article
Google Scholar
Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, et al. The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol Genet Metab. 2017;120(3):163–72.
CAS
PubMed
Article
Google Scholar
Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3(2):132–8.
CAS
PubMed
Google Scholar
Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356(9227):397–8.
PubMed
Article
Google Scholar
Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149(1):89–97.
CAS
PubMed
PubMed Central
Article
Google Scholar
Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99–109.
CAS
PubMed
Article
Google Scholar
Hahn A, Schänzer A. Long-term outcome and unmet needs in infantile-onset Pompe disease. Ann Transl Med. 2019;7(13):283.
CAS
PubMed
PubMed Central
Article
Google Scholar
Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267–88.
PubMed
PubMed Central
Article
Google Scholar
• Harlaar L, Hogrel JY, Perniconi B, Kruijshaar ME, Rizopoulos D, Taouagh N, et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology. 2019;93(19):e1756-e1767. This paper critically analyzes the long term experience with enzyme replacement therapy and how the treatment loses its benefit over a period of years, often starting at 36 months. The loss of benefits was across the board in all functional elements.
Papadimas GK, Anagnostopoulos C, Xirou S, Michelakakis H, Terzis G, Mavridou I, et al. Effect of long term enzyme replacement therapy in late onset Pompe disease: a single-centre experience. Neuromuscul Disord. 2021;31(2):91–100.
PubMed
Article
Google Scholar
•• Diaz-Manera J, Kishnani PS, Kushlaf H, Ladha S, Mozaffar T, Straub V, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20(12):1012–26. This paper reports the results of the Phase 3 randomized controlled trial of avalglucosidase alfa vs. alglucosidase alfa in treatment naive patients with LOPD. This was a true head-to-head comparison of avalglucosidase alfa, a newly designed ERT, with more M6P moeities, to alglucosidase alfa, the standard of care in LOPD since 2010. The results show that avalglucosidase alfa was non-inferior to alglucosidase alfa, but there were trends in some outcome measures that showed avalglucosidase alfa to be significantly better than alglucosidase alfa. This resulted in FDA approval of avalglucosidase alfa for treatment of all forms of Pompe Disease in 2021.
•• Schoser B, Roberts M, Byrne BJ, Sitaraman S, Jiang H, Laforêt P, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021;20(12):1027–37. This paper reports the results of the Phase 3 randomized controlled trial of cipaglucosidase alfa in combination with miglustat, a stabilizer of the enzyme, vs. alglucosidase alfa and a placebo in treatment naive patients with LOPD. This was comparison of cipaglucosidase alfa, a newly designed ERT with through clonal selection of rhGAA with CHO-cell derived M6P and bis-M6P moieties, to alglucosidase alfa, the standard of care in LOPD since 2010. The results show that avalglucosidase alfa was non-inferior to alglucosidase alfa, but there were trends in some outcome measures that showed cipaglucosidase alfa to be significantly better than alglucosidase alfa. This new drug will go up for FDA review in Fall 2022.
• Baik AD, Calafati P, Zhang X, Aaron NA, Mehra A, Moller-Tank S, et al. Cell type-selective targeted delivery of a recombinant lysosomal enzyme for enzyme therapies. Mol Ther. 2021;29(12):3512–24. A very interesting paper that describes a two-step process being developed by Regeneron for treatment of Pompe Disease. They also use a gene therapy approach to create a liver-depot to create endogenous supply of these ERT that are conjugated with monoclonal antibodies to CD63 and ITGA7 to ensure maximal delivery to muscle and cardiac tissue.
• Zhou Z, Austin GL, Shaffer R, Armstrong DD, Gentry MS. Antibody-mediated enzyme therapeutics and applications in glycogen storage diseases. Trends Mol Med. 2019;25(12):1094–109. A good review of new efforts to develop enzyme replacement therapies conjugated with monoclonal antibodies to allow better delivery to skeletal muscle and cardiac tissue.
CAS
PubMed
PubMed Central
Article
Google Scholar
Ronzitti G, Collaud F, Laforet P, Mingozzi F. Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. 2019;7(13):287.
CAS
PubMed





留言 (0)