
記住我
The N terminal section of BORIS corresponding to AA 1–258 (BORIS-N1-258) and the N terminal-truncated BORIS (BORIS-del N1-258) were cloned by the Fast Mutagenesis System in the backbone of plasmid pFN6K. Both truncated BORIS constructs were fused with 6 consecutive histidine residues (His-tag) at the C-terminus for subsequent protein purification. Single step (KRX) competent cells (Promega Corporation) were used for protein expression. Proteins were purified by Ni–NTA column chromatography and confirmed by Western blotting using the anti-His antibody (Supplemental Fig. 1A). All plasmids used in this study are listed in Supplemental Table I.
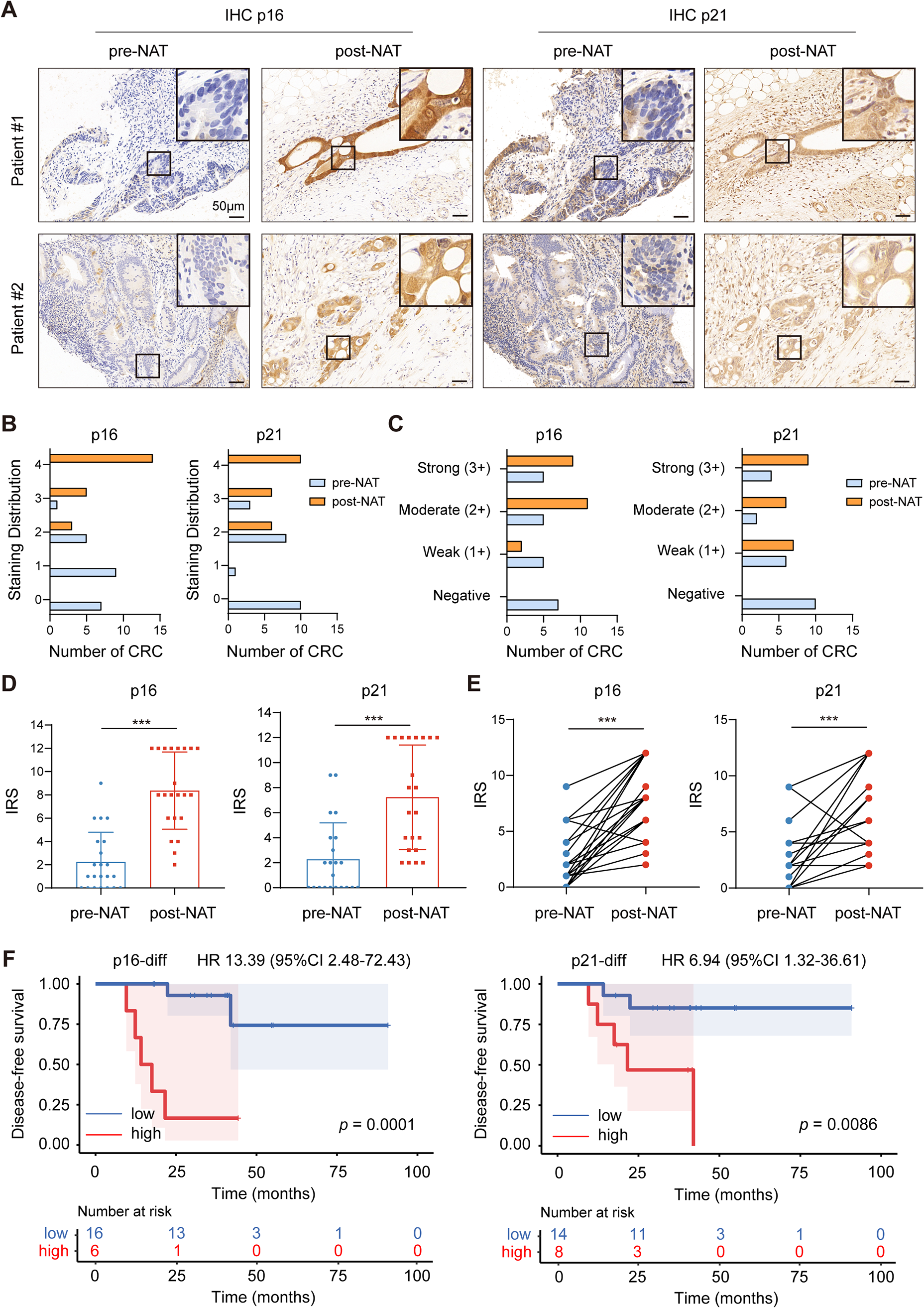
Fig. 1
Selection and characterization of the BORIS-binding peptide. (A) Procedure for the selection of BORIS-binding peptides. (B) The sequences and frequencies of the peptides enriched after elution. (C) ELISA testing the affinity of phages for the BORIS-N1-258 protein. (D) The peptide from phage clone 9 was used to determine the affinity of the interaction with BORIS-N1-258 protein by BLI. The panel shows the test of the BORIS-N1-258 protein immobilized on an SSA sensor and free peptide in solution. (E) Scrambled peptide 9 showing no affinity to BORIS-N1-258 in the BLI assay. (F) Peptide 2 showed a weak binding affinity (Kd) of 314.5 µM to BORIS-N1-258 in the BLI assay. (G) Peptide 9 immobilized on an SA sensor and free BORIS-N1-258 protein in solution. (H) The BORIS-N1-258 protein purified from HEK293 cells was used to examine the interaction with synthesized peptide 9. The test was performed by fixing peptide 9 on an SA sensor and releasing the humanized BORIS-N1-258 protein to solution in a BLI assay
Enrichment of phages from the random peptide display libraryPh.D.™-12 Phage Display Peptide Library Kit (New England Biolabs) was used for biopanning. This library contained 2 × 1011 unique 12-mer peptides linked to the N-terminus of a phage coat protein by a 4 amino acid spacer (GGGS). The library was diluted in TBS (50 mM pH 7.5 Tris–HCl, 150 mM NaCl). Microtubes coated with 100 µg/mL purified proteins in 0.1 M pH 8.6 NaHCO3 were used for the selection of phages. BORIS-del N1-258 was used in the first step of elutriation for 60 min at room temperature (RT) to remove non-specific phage clones. The pre-cleared phage library was used for further elutriations on BORIS-N1-258-coated microtubes for 60 min at room temperature and eluted with a general buffer (0.2 M pH 2.2 Glycine–HCl, 1 mg/mL BSA). The eluate enriched in BORIS-N1-258 binding phage clones was neutralized with 1 M pH 9.1 Tris–HCl. By two additional rounds of selection, phage clones enriched in BORIS-N1-258 binding clones were prepared.
Test of selected phages in ELISAThe enriched phage clones were serially diluted and spread on Luria–Bertani agar plates. Sixty clones were randomly chosen for sequencing according to the manufacturer’s protocol. Nine peptide sequences were identified from the selected clones. We counted the frequency of each peptide displayed on the selected clones and examined the interactions between BORIS-N1-258 and the phage clones by ELISAs. ELISA was carried out as follows: ELISA plates (Thermo Scientific Nunc) were coated overnight at 4 ℃ with purified His-tagged BORIS-N1-258, BORIS-del N1-258 or BSA protein (10 mg/mL in carbonate buffer, pH 9.6), washed with PBST (0.1% Tween 20 in pH 7.4 PBS) three times and blocked with 5% BSA in pH 7.4 PBS for 1 h at RT. Purified phage clones were applied to the plates in serial dilutions (diluted with PBS containing 1% BSA) and incubated for 2 h at RT. After washing three times with PBST, bound phages were detected by a horseradish peroxidase-conjugated anti-M13 antibody (GE Healthcare, 27–9421-01) followed by incubation with the TMB substrate. Reactions were quenched using 250 mM HCl, and the absorbance at 450 nm was recorded by a plate reader (BioTek, Synergy 2).
Examination of physical interaction between selected peptides and BORIS-N1-258The candidate peptides that showed potential interactions with BORIS-N1-258 and the disorganized peptide were synthesized and conjugated with biotin or FITC by China Peptides Co., Ltd. (https://chinesepeptide.chemdrug.com/sell/). Twelve tandemly connected His was synthesized and conjugated with biotin or FITC as a negative control peptide by China Peptides Co., Ltd. The peptides were stored in powder at -80 °C. The peptides were dissolved in phosphate-buffered saline and filtered through a 0.22 µm filter to remove bacteria before use. The interaction between peptides and BORIS-N1-258 was measured by Bio-layer interferometry (BLI) in the ForteBio OctetRed system. The measurement was carried out in 5 steps: initial baseline duration, loading duration, baseline duration, association duration, and dissociation duration. In loading duration, 10 nM biotinylated peptides or 1 µM BORIS-N1-258 protein were immobilized on streptavidin-coated biosensors (ForteBio). The immobilization typically reached a response level of 4 nm. Association and dissociation curves were obtained through the addition of a dilution series of BORIS-N1-258 or peptides in PBS with 0.02% Tween 20 for the indicated period of time using Octet acquisition software. The binding data were fitted using Octet analysis software.
Cell cultureH1299 cells (RRID: CVCL_0060) were cultured in RPMI 1640 medium supplemented with 10% FBS. HEK293 (RRID:CVCL_0045) and HeLa (RRID:CVCL_0030) cells were cultured in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% FBS and placed in 37 ℃ and 5% CO2 incubators. Cells were seeded in 6-well plates at 1 × 105 cells/well or in 96-well plates at 2000 cells/well for experiments. Cell viability was measured by MTT assay.
Examination of interaction between selected peptides and BORIS-N1-258 in cellsThe candidate peptides or negative control peptide fused with HIV-1 TAT sequence and conjugated with biotin were added to the cell lysate at a concentration of 25 µM and incubated for 24 h. Immunoprecipitation was performed to pull down peptides by streptavidin-conjugated magnetic beads (Cell Signaling Technology, 5947) and by BORIS primary antibody (Santa Cruz, CA, USA, sc-377085). Western blot assay was preformed after immunoprecipitation.
Detection of cell viability and apoptosis induced by BTApep-TAT in vitroCells were plated and treated with BORIS-targeted peptide (BTApep-TAT) or negative control peptide (His-TAT) at concentrations of 10 µM, 25 µM, 50 µM and 100 µM for 3 days. Cell viability was measured by MTT assay. Cell apoptosis was measured by TUNEL assay (cat. # 40306ES20, Yeasen Biotech Co., Ltd) and Caspase-Glo 3/7 assay (G8090, Promega Corporation, an affiliate of Promega (Beijing) Biotech Co., Ltd.).
RNA-sequencing analysisPeptide-treated and siRNA-silenced H1299 cells were collected after three days of treatment. The sequences of the siRNAs used in this study are listed in Supplemental Table II. RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA from triplicate treated samples was purified and subjected to RNA-sequencing analysis using the DNBSEQ-G50 platform (BGI-Shenzhen, China). The differential gene expression analysis was performed by the Dr. Tom online system (BGI-Shenzhen, China, http://biosys.bgi.com). The heatmap was generated by Sangerbox 3.0 (http://vip.sangerbox.com/home.html). The BioProject accession is PRJNA832514. A few differentially expressed genes involved in the modulation of DNA damage regulation was validated by quantitative real-time PCR (qRT-PCR).
Immunofluorescence assayBTApep-TAT and BTApep were conjugated with biotin. The biotin-conjugated peptides were added to cell culture medium. Intracellular distributions of biotin-conjugated peptides were detected by Alexa Fluor® 488 Streptavidin (Yeasen Biotech Co., Ltd., Shanghai, China) and analyzed by an ImageXpress Micro Confocal system (Molecular Devices, LLC. San Jose, CA, USA).
Quantitative real-time PCRRNA was extracted from the cell pellet using TRIzol (Thermo Fisher Scientific) and ethanol precipitation. After quantification using a Nanodrop 2000 system, equal amounts of RNA from control and experimental samples were reversely transcribed into cDNA. The expression of candidate genes was quantified by qRT-PCR using GAPDH as a reference gene. The primers used in this study were reported in our previous study and are listed in Supplemental Table III [24].
In vivo experiments in a mouse xenograft modelH1299 cells (1 × 106 cells/injection) were subcutaneously injected into the limbs of NOD/SCID/γc null (NSG) mice. In this study, all animals were male. One week after cell inoculation, BTApep-TAT/His-TAT (dissolved in PBS) was injected intraperitoneally at 16 mg/kg body weight every other day for 3 weeks. Tumor volumes were recorded every other day. The weight and volume of the tumors were recorded at the end after surgical resection. Serum was collected from each mouse and used for liver (ALT, AST, ALP, DBIL, TBIL) and kidney function (CRE, UA) analyses by Servicebio Wuhan, China (https://www.servicebio.cn/). Tumors were sliced and examined by TUNEL assay by Servicebio. All experimental protocols were approved by the licensing committee of Hangzhou Medical College, P. R. China.
Preparation of nuclear extractTwenty million (2 × 107) HeLa or H1299 cells were trypsinized, collected by centrifugation (2000 g, 5 min) and rinsed twice in PBS. Cell pellets were resuspended in a fivefold packed cell volume of hypotonic buffer (10 mM HEPES–KOH, pH 7.5 at 4 °C, 5 mM KCl, 1.5 mM MgCl2, 0.2 mM PMSF, 0.5 mM DTT), kept on ice for 10 min, and centrifuged (10 min, 1200 g). The cell pellets were resuspended in an equal volume of hypotonic buffer and disrupted in a Dounce homogenizer (20 strokes, pestle B). Subsequently, 3 M KCl was slowly added to a final concentration of 50 mM KCl, and the mixture was kept on ice for 10 min and centrifuged (3000 g, 20 min) to precipitate the nuclei. The nuclear pellets were resuspended in 2 packed nuclear volumes of low salt buffer (20 mM HEPES, pH 7.9 at 4 °C, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 0.2 mM PMSF, and 0.5 mM DTT) and added to 1 volume of high salt buffer. Nuclear proteins were extracted at 4 °C for 30 min under gentle agitation and centrifuged at 10,000 × g for 30 min at 4 °C.
In vitro assay for detecting DNA single-strand break repair (SSBR) by qRT-PCRSSBR activity was analyzed by qRT-PCR, as described previously [25]. Crude nuclear extracts were isolated, and the protein content was quantified by the Bradford assay. Twenty-four micrograms (24 µg) of nuclear protein were incubated for 30 min at 32 ℃ in a 20 µl reaction mixture that contained 45 mM HEPES–KOH, 70 mM KCl, 7.4 mM MgCl2, 0.9 mM DTT, 0.4 mM EDTA, 2 mM ATP, 20 µl each of dATP, dTTP, cCTP, and dGTP, 40 mM phosphocreatine, 2.5 µg of creatine phosphokinase, 20 µg/ml BSA, 3.4% glycerol, 2 mM NAD + , 4 µg of poly(dI/dC), and templates listed in Supplemental Fig. 2 (2 pmol DNA template A, which had a break, or template B, which contained a single nucleotide deletion, or template C, which was endogenous control DNA). The reaction was terminated by heating at 72 °C for 10 min. Two microliters of 10,000 × dilution of the reaction mixture was used as a template for qRT-PCR. The probes and primers were designed as described [25]. The final concentrations of the forward and reverse primers were 200 nM. The final concentration of either probe was 300 nM. qRT-PCR monoplex reactions were performed by annealing at 60 °C for 40 cycles. The quantity of repaired templates was calculated by comparing the Ct values of repaired templates and control template.
Fig. 2
BTApep-TAT induced DNA damage and cancer cell apoptosis. (A) Co-immunoprecipitation was performed to evaluate the interaction of BTApep-TAT-biotin with BORIS-N1-258, full length BORIS, and BORIS-del N1-258 in the cell lysates from transfected H1299 cells. (B) The level of BORIS in H1299 cells after siRNA-mediated knockdown was evaluated by BORIS antibody or the BTApep-TAT-biotin peptide. (C) Cells were incubated with graded concentrations of the peptides (25–100 µM) for three days. MTT assays and cell counting were performed to evaluate the effect of BTApep-TAT and the negative control peptide His-TAT on H1299 cells. (D) H1299 and HEK293 cells were treated with 25 µM BTApep-TAT or BTApep to examine the effect of BTApep-TAT on cancer cells and normal cells (E) Transcriptomes of H1299 cells with siBORIS knockdown or BTApep-TAT treatment were compared. The left panel shows an overlap between siBORIS knockdowns and BTApep-TAT treatment in a Venn diagram. Two siRNAs targeting BORIS were used to compare the common genes in the heatmap. (F) A bubble map showing the pathways associated with the genes common to BORIS knockdown and BTApep-TAT treatment, which are shown in Panel E. (G) Caspase 3/7 assay detected the peptide-induced apoptosis at peptide concentrations from 10 to 100 µM. (H) A TUNEL assay detected the DNA damage induced by 25 µM peptide
In vitro end-joining assayA plasmid-based assay for in vitro end joining was performed as described [26]. The end-joining reaction was performed in a final volume of 30 µl by incubating 100 ng of XhoI-digested pCMV6 plasmid (RRID:Addgene_58320) with 10 µg of nuclear extract from HeLa cells for 1 h at 25 °C in NHEJ buffer (20 mM HEPES–KOH, pH 7.5 at RT, 80 mM KCl, 10 mM MgCl2, 1 mM ATP, 1 mM DTT, 50 mM dNTP, 80 mM NaCl and protease inhibitors). The reaction was stopped with 2 µl of 0.5% SDS, 2 µl of 0.5 M EDTA, followed by 1 µl proteinase K (10 mg/mL) treatment at 37 °C for 0.5 h. Ten microliters of the samples were separated by running on a 0.7% agarose gel at 2 V/cm in 0.5 × TBE buffer. The gel was stained with SYBR Gold I (diluted 1:20,000 in 0.5X TBE) and visualized under UV light.
Fluorescence-based DNA repair assay in cellsAlternative NHEJ (RRID:Addgene_44025), total NHEJ (RRID:Addgene_44026), and DSB Repair (Homology directed repair) DNA repair report (RRID:Addgene_26475) systems were used to investigate the function of BORIS in cells [27]. BORIS-RFP plasmid with pCMV6-Entry backbone and fluorescent-based DNA repair reporter plasmids were co-transfected into HeLa cells for 24 h. Flow cytometry was used to count and analyze the cells with red and/or green fluorescence. The cells expressing only GFP or RFP were used as controls. The cells expressing both red and green fluorescence indicated that BORIS-RFP was expressed and that DNA damage was successfully repaired. The cells without red fluorescence were used to analyze spontaneous DNA repair. The percentage of cells that underwent DNA repair was determined by calculating the percentage of cells with GFP fluorescence in BORIS-RFP-transfected cells or cells without transfection (or without RFP fluorescence). The proportion of cells with GFP fluorescence was compared between cells with and without RFP fluorescence.
Detection of ADP ribosylation of BORISHeLa or H1299 cells were lysed in immunoprecipitation (IP) buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) containing protease inhibitor cocktails. The cell lysates were incubated with mouse anti-Myc (CST, 2276) or rabbit anti-poly/mono-ADP Ribose (E6F6A) monoclonal antibody (CST, 83732) on a rotary shaker at 4 °C overnight. Mouse or rabbit IgG (Santa Cruz, sc-2025 or sc2027) was used as a negative control for detecting the pull-down specificity. Protein G beads (Santa Cruz, sc-2001) or protein A beads (Santa Cruz, sc-2002) were added and incubated at room temperature for 2 h. The agarose beads were collected by centrifugation, washed five times with IP buffer according to the manufacturer’s instructions, and eluted in SDS sample buffer for the subsequent Western blotting assay. Olaparib (Selleck, S1060) was purchased from Selleck Chemicals. The instrument used for X-ray irradiation was a RAD SOURCE RS 2000pro-225 X-RAY IRRADIATOR.
In vitro ADP-ribosylation assayDynabeads™ M280 Streptavidin Beads (Invitrogen, 60,210) immobilized with 5 pmol biotin-PAR polymers (Trevigen, 4336–100-01) or empty Streptavidin beads were incubated together with 12.5 to 100 pmol of purified BORIS protein in NETN buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 2 mM EDTA, 0.5% NP-40). After incubation for 1 h at room temperature, beads were washed with NETN buffer 5 times, and bound proteins were released by adding 30 µl SDS sample buffer followed by heating at 90 °C for 10 min for the subsequent Western blotting assay.
PARP1-catelyzed in vitro poly ADP ribosylation (PARylation) assayPeptides (1–4 μg) incubated with different samples were added to 50 μL PARP1 reaction buffer (50 mM Tris–HCl at pH 7.4, 2 mM MgCl2, 200 μM NAD+), which contained 0.2 μg of recombinant PARP1 (Trevigen, 4668–100-01) and 2.5 μg of ssDNA (Sigma, D8899), and reaction was carried out at 37 °C for 30 min. Low molecular weight peptide and PARP1 protein in the reactions were separated by a 30 kDa cutoff centrifugal filter (Millipore, UFC803096). Then, SDS loading buffer was added to separate peptides or proteins and analyzed by dot blotting using an anti- ADP ribosylation (ADPr) antibody.
Antibodies and co-immunoprecipitationsThe BORIS antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, sc-377085). Streptavidin-conjugated magnetic beads (CST, 5947), Myc-Tag (CST, 2276) mouse monoclonal antibody, and poly/mono-ADP ribose (E6F6A) rabbit monoclonal antibody (CST, 83,732) were purchased from Cell Signaling Technology, (Danvers, MA). Anti-mono-ADP ribose recombinant antibody (Sigma, MABE1076) was purchased from Sigma. Anti-poly-ADP ribose monoclonal antibody (Trevigen, 4335-MC-100) was purchased from Trevigen. All antibodies used in this study are listed in Supplemental Table IV. The materials of other chemicals and reagents are listed in Supplemental Table V.
Statistical analysisAll data were obtained in a minimum of triplicates and are expressed as the mean ± standard deviation (SD). Statistical differences between the controls and treatments were evaluated by two-tailed Student’s t test. P < 0.05 was considered statistically significant.
留言 (0)