
記住我


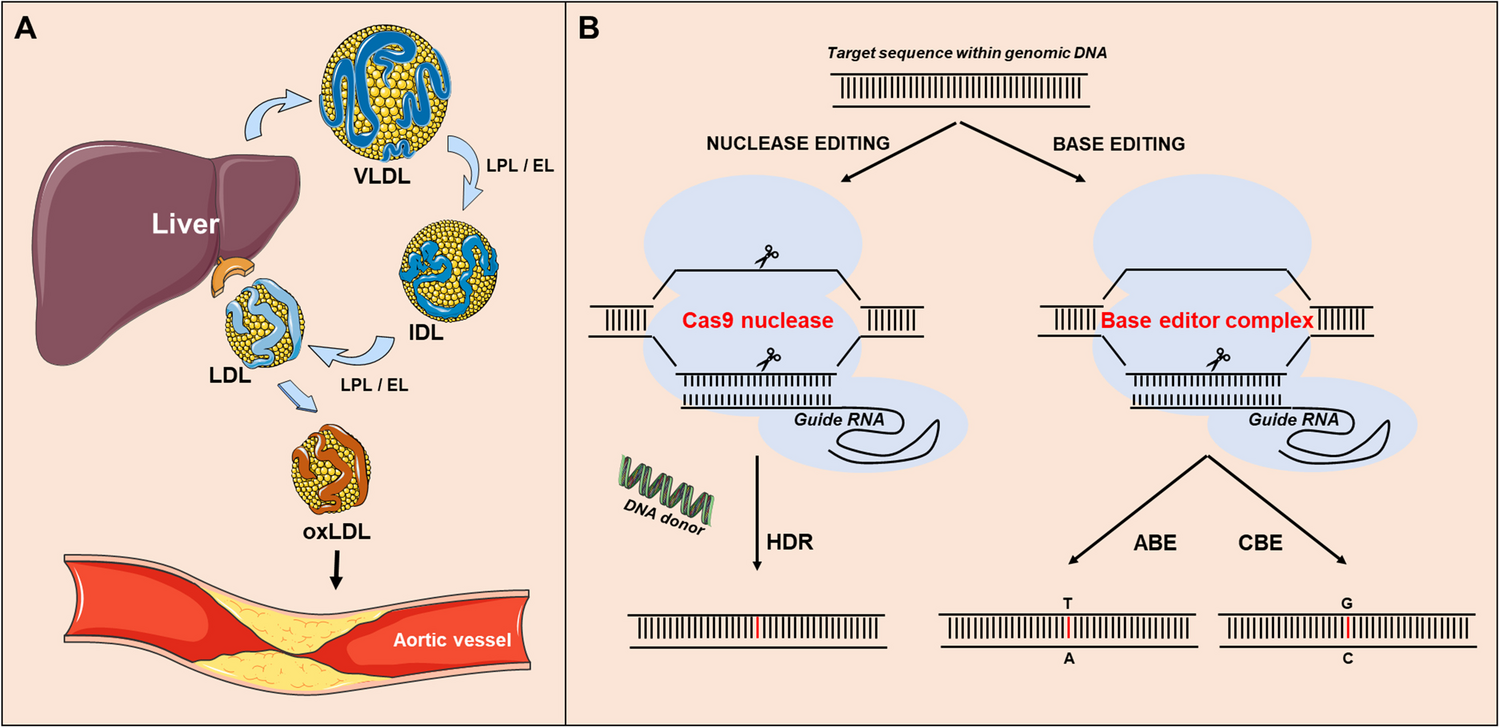



The levels of LDLc have a direct consequence on the rate and extent of development of CVD including atherosclerosis and its vascular complications. Atherosclerosis can be traced to the accumulation of LDLc in blood vessels, resulting in endothelial cell dysfunction. Coronary artery disease (CAD) is characterized by atherosclerosis formation that can lead to sudden cardiac death. Indeed, multiple clinical trials over the last decade confirmed this conclusion and quantified the large cardiovascular benefits attained upon using PCSK9i on top of maximally tolerated statins [27,28,29,30]. Furthermore, PCSK9i reduce LDLc concentrations by up to 60% and decrease the risk of major adverse cardiovascular events (MACE) in patients with acute coronary syndrome, and the absolute benefit increases with the number of metabolic risk factors [31]. The combination of statin and a PCSK9 mAb after a myocardial infarction result in favourable changes in coronary atherosclerosis consistent with stabilization and regression of the plaque burden [32]. It has recently been suggested from mouse studies that silencing both PCSK9 and LRP6, which contributes to atherosclerosis through the canonical Wnt/β-catenin pathway, may be even more effective in the treatment of this disease [33]; however, its application to patients has yet to be validated.
Monocyte infiltration into the subintimal space and its intracellular lipid accumulation are the most prominent features of atherosclerosis, which nowadays is considered as an inflammatory disease [34]. One factor leading to lipid accumulation and associated inflammation is oxidized LDL (oxLDL), resulting from the oxidation of small dense LDL in patients with high circulating LDLc. While circulating PCSK9 is primarily derived from hepatocytes [17], it is also expressed in other tissues such as small intestine, kidney, pancreas and immune system [1]. Several studies have concluded that PCSK9 expression in immune cells (e.g. dendritic cells and macrophages) [35,36,37] is mediated by oxLDL that binds the lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) leading to its enhanced expression and activation [37]. The mechanism is mediated by NF-κB a nuclear receptor responsible for expression of inflammatory genes [36, 38]. Notably, RNA sequencing analysis of mouse intimal macrophages from atherosclerotic aorta revealed that lipid-loaded plaque macrophages are not likely to drive lesional inflammation. Rather, intimal non-foamy macrophages formed the major population of cells expressing IL (interleukin)-1β and many other inflammatory transcripts in the atherosclerotic aorta [39].
Familial hypercholesterolemia (FH) is associated with low-grade systemic inflammation, a key driver of premature atherosclerosis. Clinical analysis of FH patients treated with a PCSK9 mAb demonstrated a significant reduction in inflammatory markers. The data revealed that functional PCSK9 inhibition reduces systemic inflammation and endothelial dysfunction by constraining leukocyte-endothelium interactions [40]. Thus, by preventing further cardiovascular events PCSK9i may constitute a new therapeutic approach to control the inflammatory state associated with FH. Therefore, much more work is needed to understand the origins of the inflammatory response in atherosclerotic plaques and what role(s) does PCSK9 play therein.
PCSK9 and CD36: Roles in SteatosisThe 3D-structures of PCSK9 [8, 9] revealed the existence of three distinct structural domains each playing critical roles in the regulation of PCSK9 and its intracellular traffic: a prodomain (aa 31–152), a catalytic subunit (aa 152–421), a hinge region (h; aa 422–452) and a C-terminal (aa 453–692) Cys/His-rich domain (CHRD; [12]) composed of three repeat modules (M1, M2, M3) (Fig. 1). The M2 module contains 9 out the 14 His residues found in the CHRD, which likely exert pH-dependent roles, especially at the acidic pH of endosomes. The catalytic subunit binds the EGF-A domain of the LDLR, but the trafficking of the PCSK9-LDLR complex to lysosomes for degradation is critically dependent on the CHRD (for review see [13]), especially the M2 domain [41]. Interestingly, the M1-M2-M3 modules exhibit a structural homology to the homotrimer resistin [42], a small cytokine associated with obesity and diabetes, and whose plasma levels have been linked to inflammation, cancer, atherosclerosis and cardiovascular disease [43]. It was also suggested that circulating resistin enhances the transcription of SREBP2, PCSK9 and LDLR, and in the absence of PCSK9, it could also partially reduce LDLR protein levels by an undefined mechanism [44].
Fig. 1
Schematic representation of the PCSK9-induced LDLR and CD36 degradation in endosomes/lyosomes under regular or high fat diet. (A) LDLR degradation requires the catalytic subunit of PCSK9. Under high fat diet: (B) the absence of PCSK9 results in enhanced cell surface levels of the LDLR and CD36, and accumulaion of intracellular lipid droplets (LD); and (C) cell surface CD36 is partially reduced likely due to its ability to bind the PCSK9-CHRD and to enter endosomes even in the presence of a PCSK9 mAb targeting its catalytic subunit. The various LDLR domains are emphasized, as reported [93]. For PCSK9, the prodomain, catalytic, hinge (h) and CHRD M1, M2 and M3 subdomains are also shown
So far, PCSK9 has been shown to bind and enhance the degradation of a number of not only LDLR-related receptors such as VLDLR, ApoER2 [7] and LRP1 [45], but also the fatty acid transporter and scavenger receptor CD36 [46]. The mechanism behind the PCSK9-induced enhanced degradation of CD36 is still obscure, but as for the LDLR-family members [47], it seems to implicate both extracellular and intracellular [46, 48•] activities of PCSK9. The extracellular loop of CD36 does not seem to bind the catalytic subunit of PCSK9, since incubation of PCSK9 with the mAb evolocumab did not impede the ability of the evolocumab/PCSK9 complex to bind and internalize CD36 into endosomal-like structures [48•]. This suggested that the binding domains that PCSK9 uses to interact and degrade both the LDLR and CD36 are distinct, and that CD36 may rather bind the CHRD of PCSK9 (Fig. 1), but the CHRD modules or the residues within each protein that are implicated in such binding are not yet defined. In that context, we recently reported that Pcsk9−/− mice exhibit increased expression of renal tubular CD36 and LDLR, which was associated with increased renal lipid accumulation [48•]. Furthermore, in mice fed a high fat diet (HFD), the absence of PCSK9 expression (i.e. knockout or siRNA treatment) results in a large increase in functional cell surface CD36 levels in hepatocytes and kidney cells, shown to be associated with a significant accumulation of lipid droplets and enhanced steatosis within hepatocytes [49] and renal epithelia [48•] (Fig. 1). These observations suggest that PCSK9 may play an important role in regulating both cholesterol and triglyceride levels in circulation and/or in target tissues in part via CD36. Accordingly, genetic PCSK9 deficiency further exacerbated HFD-induced renal endoplasmic reticulum (ER) stress, inflammation, and fibrosis. Amazingly, this effect is not seen in wild type mice fed a HFD and treated with a PCSK9 mAb, where the evolocumab/PCSK9/CD36 complex partially accumulates in endosomal-like structures, with little CD36 at the cell surface (Fig. 1C), thereby protecting against lipid-induced renal injury [48•]. If validated in human, these observations may have significant consequences, since our data revealed that the administration of PCSK9 mAbs may have additional clinical benefits in alleviating diet-induced renal stress and injury. Future studies should define whether intracellular and/or extracellular expression of PCSK9 act to mitigate ER stress in chronic kidney disease (CKD), and to what extent the clinically approved mAbs against PCSK9 protect against renal stress through cell surface CD36 degradation (Fig. 1C).
Since the successful introduction of PCSK9 mAb therapy to patients, there has been a rapid development focused on generating new approaches at reducing circulating PCSK9 levels and/or activity [20]. One such approach is inclisiran, a siRNA targeted against hepatic PCSK9, which has recently undergone phase III clinical trials [50]. Although this hepatocyte-targeted siRNA approach has demonstrated great efficacy in reducing circulating LDLc, its potential effects on increasing surface CD36 levels on hepatocytes and/or other tissues deserve to be investigated and compared to the mAb treatment.
PCSK9: Chaperone Functions and Transcriptional Downregulation by CaffeineAfter the discovery of PCSK9, it was soon realized that it can bind the LDLR in the ER and that such early interaction along the secretory route regulates the amount of both mature proteins that can reach the cell surface and/or be secreted [12]. These results suggested a chaperone-like role of the LDLR in facilitating the autocatalytic cleavage of proPCSK9 into mature PCSK9 and its subsequent trafficking to post-ER sites in the secretory and endocytic pathways where PCSK9 activity occurs [12]. The reverse is also true, as PCSK9 itself can act as a chaperone that enhances the exit of the LDLR from the ER [51]. Thus, the dynamic interaction of the precursor proPCSK9 and mature PCSK9 with the LDLR starts in the ER, and the mature PCSK9-LDLR complex is then targeted to the Golgi, cell surface and endosomes, ultimately leading to its degradation [12, 51, 52]. Whether such chaperone effects of PCSK9 also occurs with its other target receptors e.g. VLDLR, ApoER2, LRP1 or CD36 will need to be verified experimentally.
The clinical relevance of proPCSK9 binding to the LDLR in the ER was emphasized upon the discovery of a natural PCSK9Q152H heterozygote/homozygote variant in French Canadian families [18]. This loss-of-function (LOF) variant resulted in the inability of proPCSK9 to autocatalytically cut itself in the ER at the cleavage site Gln152↓, and hence almost complete elimination of secreted mature PCSK9 from circulation [18]. Individuals carrying this variant seem to be healthy despite a lifelong state of hypocholesterolaemia and retention of PCSK9Q152H in the ER. Intriguingly, the original observation that proPCSK9 oligomerizes in the ER [1], led to the realization that in heterozygote subjects proPCSK9Q152H can act as a dominant-negative preventing the ER-exit of the non-mutated PCSK9 derived from the wild type allele [18, 53]. The critical recognition of the autocatalytic P1 cleavage site at Gln152↓ by PCSK9 was further confirmed by site-directed mutagenesis that demonstrated that the only P1 residues potentially recognized by PCSK9 are Gln > Met > Ala > Ser > Thr ≈ Asn, and in all the above mutants proPCSK9 acted as a dominant negative preventing the exit of wild type PCSK9 from the ER [53]. In view of the original observation that proPCSK9 can retain the LDLR in the ER [51], it was possible that aside from the virtual absence of mature PCSK9 in circulation, the ER-localized proPCSK9Q152H would also retain some of the LDLR therein, resulting in lower levels of LDLR at the cell surface. However, in patients carrying PCSK9Q152H variant, the levels of LDLc were reduced by ~ 2–threefold (and not increased) [18, 19], suggesting that the effect of the loss of extracellular PCSK9 is the dominant force regulating hepatocyte cell-surface LDLR levels, compared to the LDLR retention in the ER by proPCSK9Q152H.
Chaperones ensure that the constant influx of nascent proteins entering the ER are properly folded before their exit and subsequent secretion from the cell. Intriguingly, the intracellular retention of proPCSK9Q152H did not result in an ER-storage disease [19], likely due to its ability to bind the ER-chaperone GRP94 [54, 55], which shields the ER-resident PCSK9 from the glucose-resident protein (GRP76) that acts as a sensor of the unfolded protein response (UPR) [19]. It is likely that the ER-resident GRP94 can act as an antagonist of the PCSK9-LDLR complex, preventing the intracellular route of LDLR degradation [47], thereby promoting cell-surface LDLR expression, and reducing circulating LDLc levels [54]. We surmise that therapies that block de novo synthesis of PCSK9 protein or its autocatalytic activation in the ER could result in favourable CVD outcomes.
Depletion of ER-Ca2+ results in ER stress and functional activation of SREBP-2 [54] following its processing by the membrane-bound proteases SKI-1/S1P and S2P in the cis/medial Golgi [56, 57]. Notably, both PCSK9 and LDLR mRNA levels are upregulated by the lipid regulating transcription factor SREBP2 [58,59,60]. To reduce ER stress, Lebeau et al. screened for compounds that enhance ER-entry of Ca2+ and hence augment the stabilization of GRP76 and retention of SREBP-2 in the ER. Amazingly, caffeine and some of its xanthine derivatives were shown to increase hepatic ER Ca2+ levels, and effectively block de novo synthesis of PCSK9, and reduce its circulating levels in vitro, in vivo, and in healthy volunteers [61•]. This resulted in increased expression of the cell surface LDLR protein and clearance of LDLc [61•], reinforcing the notion that loss of extracellular PCSK9 effectively enhances hepatocyte cell-surface LDLR protein levels [62]. These findings delineate a mechanism by which ER Ca2+ and its modulators e.g. caffeine, can affect the expression and activity of proteins that play a pivotal role in CVD.
PCSK9 in Viral InfectionsGenetic [63, 64] or adenoviral overexpression [65, 66] of the GOF PCSK9D374Y rapidly and effectively decreased liver LDLR levels avoiding the need for Ldlr−/− mouse models to study diseases. This method is now regularly used to generate enhanced atherosclerosis even under a regular diet [67]. A variety of enveloped RNA viruses exploit the proprotein convertases Furin and/or SKI-1/S1P for the regulation of viral infectivity and spread to multiple tissues [68]. However, the role of the proprotein convertase PCSK9 in the regulation of viral infection and its consequential damages is just starting to be appreciated.
Hepatitis C virus (HCV) associates with lipoproteins to form “lipoviral particles” (LVPs) that can facilitate viral entry into hepatocytes. Early on, it was observed that hepatitis C patients infected with a genotype 3 virus (HCV-G3) exhibited lower PCSK9 and LDL in circulation, implying upregulated LDLR activity in these patients [69]. This was the first indication that PCSK9 was the strongest negative predictor of LVP levels and hence PCSK9 may be protective against some form of HCV infection. Some of the key HCV entry receptors, including LDLR, VLDLR, SRB1 and CD81 [70] are degraded in the presence of PCSK9, also suggesting that PCSK9 may impede some cases of HCV infection [71].
Dyslipidaemia is highly prevalent in patients living with HIV and contributes to their increased risk of developing CVD, likely due to the HIV infection itself and certain types of antiretroviral therapy (ART). Indeed, lipid modification is a challenge in patients with living with HIV. Circulating PCSK9 levels were reported to be ~ 65% higher in HIV patients undergoing ART therapy, and that this increase is related to endothelial dysfunction [72], a pathology rapidly reversed by evolocumab treatment [73]. Atherosclerosis in the setting of HIV infection includes increased vascular inflammation, worsened endothelial function, and a predominance of non-calcified plaque. Since 2018, a randomized, placebo-controlled phase-III clinical trial is ongoing to evaluate the effects of alirocumab (a PCSK9i) for the reversal of coronary endothelial impairment and reducing arterial inflammation in HIV patients (NCT03207945).
Early observational studies have suggested that prior use of statins is associated with a ~ 50% reduced risk of adverse clinical outcomes in patients with COVID-19 [74], suggesting that LDLc and inflammation are key to some of the late manifestation of this deadly SARS-CoV-2 infection. This conclusion was later supported by a large meta-analysis that suggested that statins significantly reduce the risk of adverse outcomes of COVID-19 [75]. While the convertase Furin plays a key role in SARS-CoV-2 infection by activating its spike glycoprotein [76], no association between plasma PCSK9 levels and the severity of COVID-19 infection and associated inflammation is yet available. Therefore, randomized clinical trials are urgently needed to establish the usefulness of routine combined use of statins and PCSK9 inhibitors in the treatment of SARS-CoV-2 or other coronavirus infections and their associated cardiovascular complications.
In contrast, dengue virus (DENV) infection clearly showed a benefit for PCSK9i treatment, which may account for the lack of antiviral efficacy of statins [77] that enhance the expression of PCSK9 [58]. DENV is a single positive-stranded RNA virus transmitted to human by the Aedes aegypti mosquitoes resulting in ~ 25,000 deaths/year, mostly children from southeast Asia [78]. We showed that DENV infection induces the expression of PCSK9 mRNA and protein in hepatocytes [79], thereby reducing cell surface levels of LDLR and LDLc uptake, resulting in low levels of cholesterol in the ER and consequently enhanced de novo cholesterol synthesis by the SREBP-2 pathway [80]. Such increased cholesterol levels in the ER cause a significant reduction in the antiviral type I interferon (IFN) response in the host liver hepatocytes due to the cholesterol-induced suppression of the phosphorylation and activation of STING. This unexpected role of PCSK9 in enhancing DENV pathogenesis and cholesterol-dependent viral packaging was supported using alirocumab, which results in higher LDLR levels and lower viremia, as well as enhanced expression of antiviral IFN-response genes [79]. In the future, double-blind clinical studies would be needed to validate and support the use of PCSK9i in the treatment of DENV infections.
PCSK9 in Cancer/MetastasisBased on Northern blot analyses, the first report on PCSK9 revealed that its mRNA is well expressed in the rat immune system (thymus and spleen) and in human colon carcinoma Lovo-C5 cells [1]. In mouse, single cell transcriptomics analysis revealed that within the thymus PCSK9 mRNA is weakly expressed in immature T-cells (https://tabula-muris.ds.czbiohub.org/). However, human PCSK9 mRNA (https://www.proteinatlas.org/ENSG00000169174-PCSK9/immune+cell) is barely detectable in immune cells. Since circulating PCSK9 is predominantly derived from hepatocytes [17, 81] it was possible that circulating, rather than locally expressed, PCSK9 may positively or negatively regulate immune functions. Interestingly, administration of a PCSK9i mAb and atorvastatin to healthy monkeys did not result in immunosuppression as measured by T-cell dependent antibody responses, natural killer cell activity, immunophenotype or delayed type hypersensitivity [82]. In contrast, it was recently reported that in human FH patients T-regulatory cell activation was enhanced by alirocumab treatment, which also elevated the plasma levels of the anti-inflammatory IL-10 and lowered circulating pro-inflammatory cytokines [40]. These data suggest that PCSK9 suppresses some immune functions.
The mechanistic connection of circulating PCSK9 to the negative regulation of immune function remained obscure until 2020, when a report appeared by Liu et al. suggesting that PCSK9 may downregulate the protein levels of the major histocompatibility type-I receptor (MHC-I), by enhancing its lysosomal degradation, similar to but independent from, the LDLR [83••]. Furthermore, the use of PCSK9i also enhanced the efficacy of immune therapy targeted at the checkpoint protein PD-1 [83••]. The connection of PCSK9 to cancer was further entrenched when it was realized that PCSK9 can also reduce the antitumor activity of the CD8 + T cell receptor via its ability to enhance the degradation of the LDLR that dimerizes with the CD8 + T-cell receptor [84]. Thus, genetic deletion or pharmacological inhibition of PCSK9 can enhance the antitumor activity of CD8 + T cells by alleviating the suppressive effect of PCSK9 on CD8 + T cells, and consequently result in inhibition of tumor progression. By reducing tissue inflammatory response, intra-tumoral immune cell infiltration and tumor progression, these seminal reports emphasized the clinical relevance of the co-administration of PCSK9i to potentiate immune checkpoint therapy or chemotherapy in the fight against cancer and its associated metastasis [85, 86].
Interestingly, Liu et al. reported that the M2 domain of PCSK9 binds the N-terminal a1-loop of the MHC-I H2-K and HLA-A2 mouse and human proteins, at the motifs R68-Y-E70 and R68-M-E70, respectively. Alignment of the various human and mouse MHC-I proteins around the R-X-E motif revealed that the X residue can be M, G, E or V in human and Y, F, M or D in mouse MHC-I proteins, respectively (not shown). Thus, while X is a variable, it seems to prefer small or hydrophobic residues but not positively ones (R or K). It will be exciting to identify the exact residues in the M2 domain of the PCSK9-CHRD that interact with the R-X-E motif in MHC-I receptors and whether the binding of PCSK9 to MHC-I favours or rather interferes with its ability to enhance the degradation of the LDLR also requiring the M2 domain [41].
PCSK9 in MHC-II RegulationPCSK9 is expressed in extra-hepatic tissues such as small intestine, lung, kidney and pancreas [1]. We recently generated tissue-specific knockout mice in the small intestine [87] and β-cells of the pancreas [88•]. Unfortunately, we failed to highlight a critical activity of extra-hepatic PCSK9 on the metabolism of circulating lipids, suggesting a role beyond lipid metabolism in these tissues. In agreement, PCSK9 expression increased locally in endothelial and smooth muscle cells under inflammatory conditions associated with atherosclerosis, such as exposure to oxLDL and TNF-α [38]. Furthermore, adenovirus-shPCSK9 silencing of PCSK9 expression in the inflamed colon of a rat model of intestinal bowel disease (IBD) resulted in decreased Toll-like receptor 4 (TLR4) and reduced NF-κB nuclear translocation, suggesting that PCSK9 is proinflammatory by positively regulating the TLR4/NF-κB pathway [89].
PCSK9 is known to enhance the lysosomal degradation of at least 6 transmembrane receptors (i.e. LDLR, ApoER2, VLDLR, LRP-1, CD36 and MHC-I) [5, 7, 45, 46, 83••]. TLRs are involved in innate immune cell activation, and the transcription of TLR4 is regulated by PCSK9 [89]. MHC class II (MHC-II) receptors are expressed on a limited subset of somatic cells known as professional antigen-presenting cells (APC) [90]. We hypothesize that PCSK9 could also regulate one or more receptors at the cell surface of innate immune cells, such APCs, that stimulate T cells via the presentation of foreign antigens through MHC-II. However, Liu et al. reported that while PCSK9 enhances the degradation of MHC-I proteins, it seemingly did not affect MHC-II labelling in B16F10 melanoma cells [83••]. Since these cells express low levels of MHC-II under regular culture conditions, but much higher ones upon exposure to γ-interferon [91], these authors may have missed a key regulation of MHC-II by PCSK9, possibly through binding of the PCSK9 M2 domain to an R-X-E motif in MHC-II, as it did for MHC-I [83••].
The quaternary structure of MHC-II is composed of a heterodimer of a- and β-chains [90]. Accordingly, we searched for exposed R-X-E motifs in the primary and secondary sequences of the α and β chains of human MHC-II, so called HLA-DM, -DQ, -DP, -DO and –DR. Indeed, we found R-X-E motifs in the α chain of HLA-DR and HLA-DO, and in the β chain of HLA-DQ and HLA-DR (Fig. 2). Interestingly, the predicted 3D structure (https://www.uniprot.org/) revealed that these R-X-E motifs in MHC-II are in solvent exposed loops, supporting a possible interaction between PCSK9 and MHC-II (Fig. 2). In agreement, PCSK9 was reported to play a key role in the induction of CD36 (a PCSK9 target [46]) and HLA-DR expression at the cell surface of dendritic cells activated by oxLDL [35]. Could PCSK9 induce the mRNA expression of some of its targets (e.g. CD36 and HLA-DR) and yet limit their protein expression by targeting them to lysosomal degradation? Finally, in view of the high expression of PCSK9 in the small intestine and lung and the presence of R-X-E motifs in some MHC-II chains (Fig. 2), we propose that PCSK9 might be implicated in inflammatory diseases that involve MHC-II, such as food allergy and asthma [90]. Future experiments are needed to support or not the possible implication of PCSK9 in allergic diseases.
Fig. 2
Potential solvent exposed R-X-E motifs in MHC-II (A) α- and (B) β-chains both expressed on human chromosome 6. The predicted 3D structures are shown, and the variable positions of the framed R-X-E motifs are emphasized
留言 (0)