
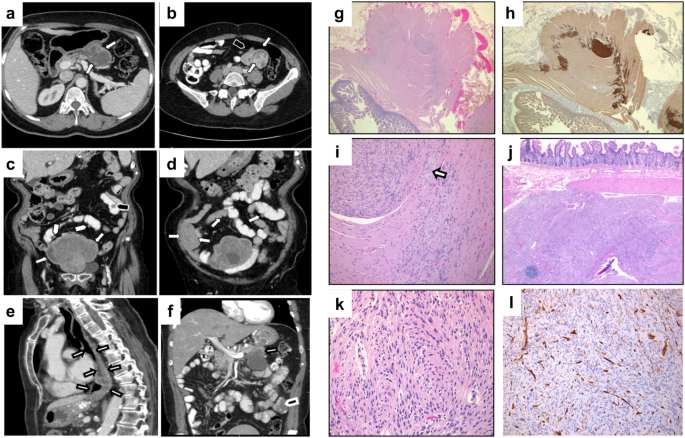
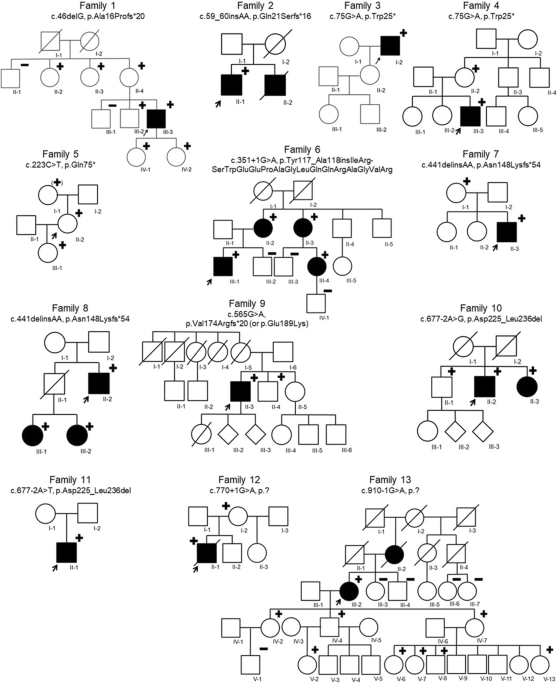
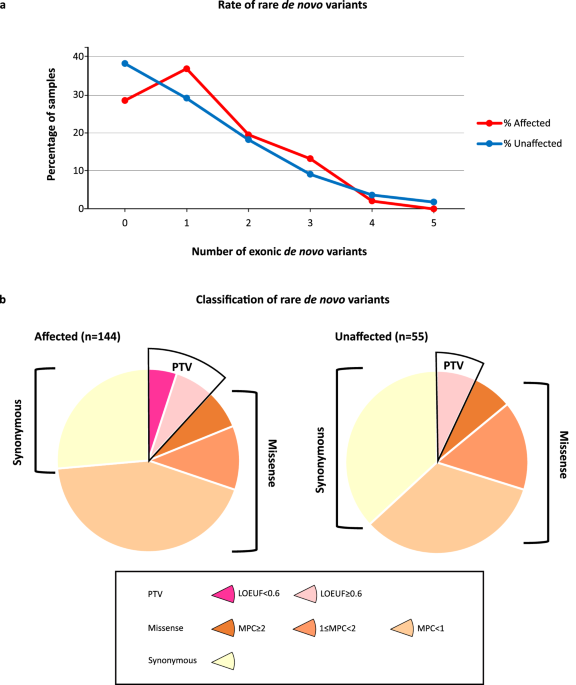
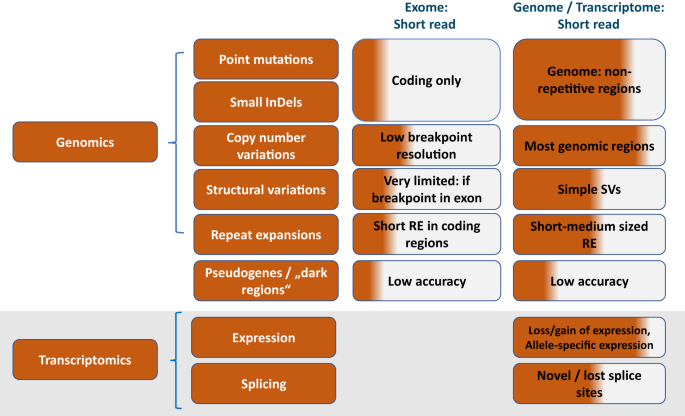
記住我


We initially retrieved 3103 genes listed as AR or XL in at least one of the four databases OMIM, CDG, ClinGen, DDG2P. For final data evaluation we excluded 45 genes that were no longer considered recessive, and remained with 3058 genes, of which ~80% showed consensus recessive/X-linked inheritance (Fig. 1a and Supplementary Tables 1a, b, and 2). Although pLI-scores, which indicate the probability of intolerance to loss-of-function, may be used to discern between recessive and dominant genes with conflicting information27, we found that this parameter is generally not accurately categorizing well-established genes (Supplementary Table 1a). Clinical categorization revealed that 1990 of the remaining 3,058 genes affected neurological (1675) and/or musculoskeletal (1082) systems (Fig. 1b and Supplementary Table 1a). Four non-coding mRNA (MIR2861, RMRP, RNU4ATAC, SNORD118), one gastrointestinal (PERCC1), and three immunologic genes (IGHM, IGKC, TRAC) were not captured with standard exome kits. Since exome sequencing has limitations in detecting non-coding repeat-expansions or variants in genes that have a paralog or pseudogene, FMR1, SMN1, GBA, HBA1, HBA2, or CYP21A2 could not reliably be assessed from the NGS data. Although CYP21A2 is known to have a high carrier frequency in a middle European population28, it is not NDD-related, usually detected by biochemical new-born screening and well treatable. Since SMN1 and FMR1 represent the most common recessive severe muscle disorder and intellectual disability disease gene, respectively, we complemented the exome sequencing data of 3045 genes (including FMR1 sequence variants) by targeted testing for these two genes and thus analyzed a total of 3046 AR/XL genes, of which 1009 were annotated as definitive NDD genes in the sysNDD database, which includes genes causing developmental delay, intellectual disability and autism spectrum disorder. Despite improvement of gene capturing by newer exome kits, all methods applied in this study showed on average a 20-fold coverage of at least 90% of the coding region in more than 96% of all targeted recessive genes as well as of NDD genes (Supplementary Table 1a and Supplementary Fig. 1).
Fig. 1: Genes and variant distributions.
a Venn plot shows numbers of 3,058 AR/XL genes common or distinct in four databases, including the Online Mendelian Inheritance in Man (OMIM), the Clinical Genomic Database (CGD), the Development Disorder Genotype - Phenotype Database (DDG2P), and the Clinical Genome Resource (ClinGen). b Venn plot shows numbers of 3058 AR/XL genes in different manifestation categories according to the CGD manifestation category definition. c Bar plot shows distributions of the percentages of the filtered variants with (ClinVar) and without ClinVar annotation (non-ClinVar) according to their approximate levels of pathogenicity scale. P/LP: pathogenic/likely pathogenic; VUS: variant of uncertain significance; B/LB: benign/likely benign. d Density plots depict distributions of the filtered ClinVar pathogenic/likely pathogenic (P/LP, dark red), ClinVar conflicting with 75%P/LP entries (Conflicting_75%P/LP, red), ClinVar variants of uncertain significance (VUS, yellow), and ClinVar benign/likely benign (B/LB, light blue) missense variants according to VIPUR score (left), CADD score (middle), and the percentage of deleterious predictions of eight conventional in silico prediction tools, including SIFT, PolyPhen2, LRT, MutationTaster, MutationAccessor, FATHMM, PROVEAN, and M.CAP (right). P-values by Welch t-test show the significant difference of the distribution between P/LP and VUS/Conflicting/B/LB (dark red letters) and between Conflicting_75%P/LP and VUS/Conflicting/B/LB (red letters). Red dashed lines indicate cut-offs (VIPUR ≥ 0.85, CADD score ≥ 20, %deleterious predictions ≥ 85%) for “high stringency” missense variants (upper panel). Likewise, density plots depict distributions of the filtered non-ClinVar missense variants (lower panel).
Variant pathogenicity assessmentFrom the exome data of 3045 genes, we obtained ~70,000 different variants from 700, mostly European individuals (Supplementary Fig. 2), which we separated in ~43,000 (61%) ClinVar and ~27,000 (39%) non-ClinVar variants (Fig. 1c, Supplementary Fig. 3). Among the ClinVar variants, we found 791 P/LP, and another 86 variants with conflicting annotations with at least 75% of entries indicating P/LP. Of these, ~56% and ~95%, respectively, had entries in HGMD, with ~90% each classified as disease-causing mutation (DM) (Supplementary Fig. 3).
Using the ClinVar missense variants as benchmark dataset, the default VIPUR score cut-off of 0.5 to predict deleteriousness29 resulted in a specificity of 66% and a sensitivity of 78% (Fig. 1d). The support-vector-machine (SVM) model combining VIPUR and sequence-based prediction scores improved specificity to 99%, but limited sensitivity to 7%. Assessing various combinations of thresholds, we found that the cut-offs of VIPUR score ≥ 0.85, CADD score ≥ 20 and ≥ 85% of other sequence predictions being deleterious (Fig. 1d), yielded a 97% specificity and increased sensitivity to 24%. Applying these thresholds to our rare (minor allele frequency (MAF) < 2%) ~17,000 different non-ClinVar missense variants, which account for ~97% of the huge amount of VUS (Supplementary Fig. 3), we obtained 402 variants, referred to as non-ClinVar “high stringency”. Additionally, we found 1083 truncating and 346 in-frame non-ClinVar variants, of which 0.05% and 0.73% were HGMD-DM, respectively (Supplementary Fig. 3). From these 1831 non-ClinVar variants we excluded 44 with a gnomAD or internal MAF > 5% or > 2 homozygous/hemizygous alleles in gnomAD. After exclusion of variants in 45 genes that later were no longer considered recessive, we obtained for the 700 individuals 3674 variants in 3046 genes, which were classified into 16 variant classification groups according to evidence levels of pathogenicity (Supplementary Fig. 3).
Notably, gnomAD all population MAFs were absent or below 0.5% (majority below 0.1%) in all ClinVar P/LP variants in genes annotated to the SysNDD database as definitive NDD (Supplementary Fig. 4).
Ethnical distribution and consanguinity in our cohortGenetic ancestry was estimated using a projection Procrustes analysis tool, LASER, showing that 519 (74.1%) individuals clustered with the reference European populations; 117 (16.7%) and 54 (7.7%) clustered with the reference Central/South Asian and Middle East populations, and 10 (1.4%) clustered with other populations, respectively (Supplementary Fig. 2). Estimation by runs of homozygosity indicated in our cohort 23 (6.6%) consanguineous, 293 (83.7%) nonconsanguineous couples, and 34 (9.7%) couples with uncertain relationship (Supplementary Table 4a).
Analysis of special disease alleles in FMR1 and SMN1We identified two mothers positive for an FMR1 premutation (67 and 134 CGG repeats, respectively), indicating a female carrier frequency of 1/175. In only one of the latter the transmission of a full mutation to an affected boy explained his NDD, which was not diagnosed previous to this carrier screening study. Based on exome data we identified 36 potential heterozygous carriers for the recurrent SMN1 ex7/8 deletion, of which 13 were confirmed by MLPA, accounting for a true carrier frequency of 1/54 despite any affected index cases (Supplementary Fig. 5).
Final number of index cases with diagnosesIn addition to the 142 index cases initially diagnosed through an extensive diagnostic workup, five were diagnosed retrospectively through our carrier screening due to inclusion of novel disease genes (MTX2, NARS1, NHLRC2, and TRAPPC4), or FMR1 screening (Supplementary Table 4b). The distribution of inheritance pattern and consanguinity of the total of 147 cases are detailed in Supplementary Table 4a.
Overall carrier frequencies, at-risk couples and NDD risk-reduction potentialThe distribution of clinical categorizations of genes showed neither a specific pattern in genes for which we found P/LP variants in individuals nor for genes with at-risk constellations in couples (Supplementary Fig. 6). Since variant classification groups 15 and 16 (non-ClinVar_non-HGMD non-canonical splice and protein length alterations) included many apparently benign variants, we did not consider them for further analysis. Accordingly, we found a total carrier frequency of up to 96.4% for at least one P/LP variant and a median of 4 P/LP variants per individual (range:0–12, Fig. 4, Supplementary Table 3a), which remained unchanged considering only autosomal genes (Supplementary Table 3b). While the majority of genes in which we found (likely) pathogenic variants showed frequencies ≤ 2%, 14 had frequencies > 2%, with HFE being most frequent (27.3%, Fig. 2). The mean of carrier frequencies for the definitive SysNDD genes was significantly lower than that for the other genes (0.28% vs. 0.40%, respectively, p = 0.007, Supplementary Table 1a), which is probably explained by natural selection against variants causing NDD. Considering established recessive SysNDD genes, top-ranked genes (frequency > 1%) included PAH, KIAA0586, PMM2, DHCR7, and MCCC2 (Supplementary Table 1a), most of which exert biochemical phenotypes, with PAH, PMM2 and DHCR7 being treatable. We also observed that generally the mean of carrier frequencies of autosomal genes found in at-risk constellation was significantly higher than that of genes not found in at-risk constellation, which might be due to the inherent cohort bias (p = 0.0238) (Fig. 3a). However, within the genes found in at-risk constellation the mean of carrier frequencies showed a trend towards lower frequencies in consanguineous couples compared to nonconsanguineous couples (p = 0.0948) (Fig. 3b).
Fig. 2: Carrier frequencies of recessive and X-linked genes observed in the 700 parental samples with indication of at-risk and transmitted at-risk genes and respective variant classification group distributions.
Carrier frequencies of recessive (a) and X-linked genes (b). Carrier frequency was calculated as the percentage of the number of individuals carrying variants of different pathogenicity groups in each gene; on the right y-axis, a descriptive proportion was given. From top to bottom, the genes were sorted according to their carrier frequencies, as well as alphabetically wherever equal. Black letters indicate genes with at least one at-risk parental couple, red for genes with at least one at-risk consanguineous parental couple, bold for genes with at least one at-risk homozygous variant, and boxed for genes with at least one couple having transmitted both at-risk genotypes. Grey letters depict genes with > 1% carrier frequencies that were not found in an at-risk constellation. Stars indicate genes with more than one variant identified in at least one individual. Pie size correlates with the number of different variants in individual genes. The carrier frequencies were calculated among the 700 healthy individuals (350 parental couples) for 3046 recessive genes. The detected variants were shown for 14 variant classification groups according to their levels of pathogenicity, which include ClinVar pathogenic/likely pathogenic (P/LP) (CV-P/LP, in dark pink), ClinVar conflicting with 75% P/LP entries (CV-conflicting_75%P/LP, in pink), both stratified according to disease-causing mutation (DM) classification in the Human Gene Mutation Database (DM, non-HGMD, or HGMD-non-DM). The next groups include ClinVar variants with zero golden stars (in light pink), non-ClinVar with disease-causing mutation classification in the Human Gene Mutation Database (non-CV_HGMD-DM, in light blue) sub-categorized into variant functional classes including truncating, high stringency missense, or protein-length alteration, Non-CV_non-HGMD (blue) variants were sub-categorized according to variant functional classes including truncating, or high stringency missense.
Fig. 3: Effects of at-risk and consanguinity status on gene carrier frequencies.
Considering variant classification groups 1–14, a Density plots show a significant difference between the mean of carrier frequency of genes that were at-risk and that of genes that were not at-risk according to their gene carrier frequencies. b Among the genes found in at-risk constellation, the mean of carrier frequency showed a trend towards lower frequencies in the consanguineous as compared to the nonconsanguineous group.
The real at-risk-couple frequency of 2.3–14.9% for autosomal recessive genes (3.1–19.1% with X-linked genes) was higher than those estimated by random virtual mating (0.5–9.4%), but became similar upon excluding recessive disease alleles found as diagnoses for the affected children (0.6–10.0%) (Supplementary Table 3a, b, Supplementary Fig. 7).
For autosomal recessive genes, up to 52 couples were at-risk for at least one gene. Of these couples, 21 (40.4%) harbored NDD at-risk genes, 27 (51.9%) non-NDD genes, and 4 (7.7%) both NDD and non-NDD genes (Supplementary Table 4b). Within these 52 at-risk couples (14.9% of all couples), consanguinity was enriched (13/52 = 25.0% vs. 10/272 = 3.7% in not-at-risk couples; p = 0.0001) (Supplementary Table 4b). Consanguinity was also significantly enriched in couples at-risk for > 1 autosomal gene (4/13 = 30.8% vs. 0/39 = 0%; p = 0.0026) (Supplementary Table 4b). 19 of the affected children inherited the at-risk alleles from the 25 at-risk NDD genes that explained their NDDs (Supplementary Table 4). This accounted for an autosomal risk-reduction potential for NDDs of 19/350 = 5.4%.
For X-linked genes, we identified up to 20 heterozygous female carriers equaling 5.7% at-risk couples. Of these, 13 (65%) concerned NDD genes, and 7 (35%) non-NDD genes. Eight of the 13 at-risk NDD alleles were transmitted to their children, including one girl who inherited a pathogenic variant in MECP2 from her mother, corresponding to an X-linked risk-reduction potential of 2.3% (Supplementary Table 3c).
Considering only the ClinVar and HGMD concordant P/LP variants for both AR and XL genes, the carrier and the at-risk-couple frequencies decreased by 44.8% and 83.6%, respectively (Fig. 4, Supplementary Table 3a). Additionally, removal of unreviewed ClinVar P/LP variants (zero golden stars) would decrease the risk-reduction potential by 14.3% from 7.7% to 6.6% through the exclusion of pathogenic variants in four genes (CRADD, DPYS, TRAPPC9, and UFC1) transmitted to the children of four couples (Supplementary Tables 4b, 5).
Fig. 4: Summary of carrier testing.
Results of carrier testing were summarized for number of detected variants, number of affected genes, and frequency of heterozygous carriers, at-risk couples, and risk-reduction potential according to the various pathogenicity thresholds, where a less conservative variant classification group is added stepwise as described for groups 1–14 in the legend of Fig. 2. It also includes the classification groups 15 (non-ClinVar_non-HGMD_non-canonical_splice) and 16 (non-ClinVar_non-HGMD_protein_length_alteration) which were not considered for further assessment. Overall, the carrier, and at-risk-couple frequencies showed the steepest increase upon inclusion of CV-P/LP_non-HGMD variants and the HGMD-DM, ClinVar conflicting variants with a high proportion of P/LP entries, as well as upon inclusion of previously unreported truncating variants. The risk-reduction potential showed a sharp increase upon inclusion of CV-P/LP_non-HGMD, CV-P/LP_HGMD-DM with zero golden stars, and non-CV_non-HGMD truncating variants.
Regarding the 4-Tier analysis scheme proposed by the ACMG committee for carrier screening27, we found that, based on carrier frequencies identified in our study, only when Tier-4 was applied, considerable increase of NDD risk-reduction potential not only for consanguineous (from none in Tier-2 to up to 43.5% in Tier-4, and up to ~10-fold from Tier-3 to Tier-4) but also for nonconsanguineous couples (from none in Tier-2 to up to 5.1% in Tier-4, and up to ~2.6-fold from Tier-3 to Tier-4) was achieved (Fig. 5). This also holds true for the at-risk-couple frequencies, in both consanguineous couples (up to ~3.2-fold increase by Tier-4 vs. Tier-2) and nonconsanguineous couples (up to ~1.5-fold increase by Tier-4 vs. Tier-2) (Fig. 5).
Fig. 5: Magnitude of clinical utility of ACMG-based 4-Tier carrier screening.
Frequency of at-risk couples and risk-reduction potential for NDDs according to a stepwise addition of the variant classification groups (1–14) are shown for nonconsanguineous and consanguineous couples for carrier screening for each of the 4-tiers recommended by the American College of Medical Genetics and Genomics (ACMG). In this carrier screening, Tier-1 includes screening of CFTR, SMN1 and medically and family-based risk genes, Tier-2 includes Tier-1 plus genes with carrier frequency ≥ 1/100, Tier-3 includes Tier-2 plus genes with carrier frequency ≥ 1/200 as well as X-linked conditions, and Tier-4 includes Tier-3 plus genes with carrier frequency < 1/200.
As expected, we found an enrichment of de novo pathogenic variants that explained the phenotype of the children in not-at-risk-couples compared to at-risk-couples (81/256 = 31.6% vs. 14/94 = 14.9%; p = 0.0017) (Supplementary Table 4b). There was no significant difference in paternal age between the de novo vs. inherited diagnoses (Supplementary Fig. 8). Finally, 203 (58.0%) couples remained without a diagnosis for their affected children, with no significant difference between the consanguineous and nonconsanguineous couples (12/23 = 52.2% vs. 172/293 = 58.7%, respectively; p = 0.66).
The NDD at-risk status of 16 (4.6%) couples remained undetected by ECS since the inherited causative variants of their affected children did not pass the filtering criteria (Supplementary Table 6). In addition, the risk for the recessive disorder in affected children was undetectable by ECS in 2 (0.6%) de novo hemizygous variants, 3 (0.9%) compound heterozygous inherited and de novo variants, and 3 (0.9%) inherited hemizygous CNVs or compound heterozygous inherited CNVs and sequence variants (Supplementary Table 4a).
留言 (0)