Cells and reagents
Human colon cancer cells HCT116 (CCL-247) were acquired from ATCC (Manassas, VA, USA). Additional cell lines were bought from ECACC (LS174t, Lovo). Cells were authenticated by Short-Tandem Repeat profiling (DSMZ, Braunschweig, Germany). Cell counting was performed using TTC Casy cell counter (OLS, Bremen, Germany). All reagents were purchased from Sigma-Aldrich (Bornem, Belgium) except when mentioned otherwise. Antibodies were purchased from Santa Cruz Technologies: HSC70 (sc-7298), phospho-cofilin Ser 3 (sc-12912); Sigma-Aldrich: PALD1; Cell Signaling: SSH1 (#13578), phospho-AKT Ser473 (#4060), akt (#9272), mTOR (#2983), phospho-mTOR Ser 2481 (#2974), cleaved-caspase-3 (clone 5A1E, #9664); or Dako: KI67 (M7240).
Cell culture
HCT116 were cultured in McCoy 5 A supplemented with 10% foetal bovine serum (FBS). LS174T were maintained in Minimum Essential Medium (MEM) supplemented with 2 mM L-glutamine, 10% FBS and non-essential amino acids (Gibco #111040-085). Lovo were maintained in Ham’s F12 supplemented with 10% FBS and 2mM L-glutamine. Cells were cultured in a 37 °C, 5% CO2 incubator. Cells were used between passages 1 and 10 and checked monthly for mycoplasma.
Plasmid transduction
Plasmids were transduced using lentiviral vectors in the GIGA institute viral vector facility. The sequences of shRNA #312 and #947 were TCATGGTCTCGCTGACAGT and ATCACAACAGGGGCCACCT, respectively. The irrelevant shRNA vector (SMARTvector lentiviral control) was purchased from Dharmacon (Lafayette, CO, USA).
Immunohistochemistry
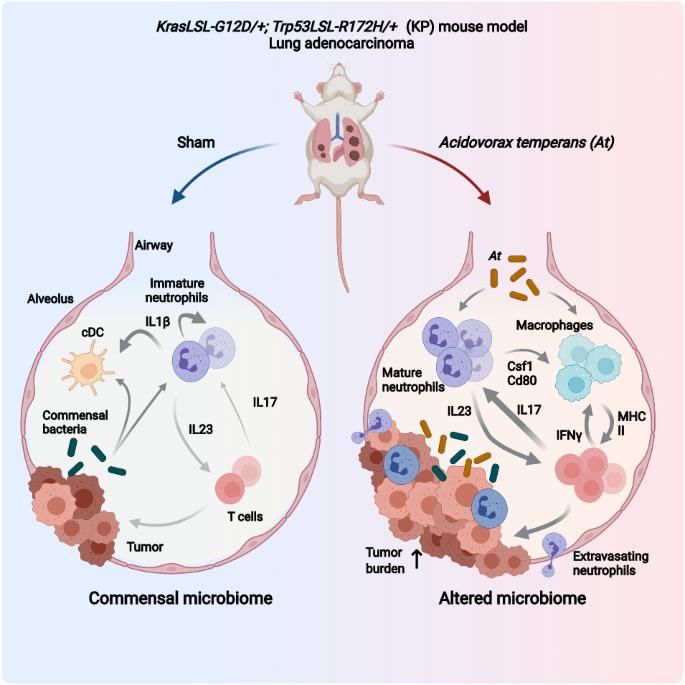
Primary tumours (n = 28) were obtained from our institution Biobank, as formalin-fixed, paraffin-embedded tissue blocks. Tissue sections were stained as described previously [33]. Based on a serial dilution, the selected optimal paladin primary antibody dilution was 1:150. Tissue sections incubated with no primary antibody showed no reactivity. Immunohistochemistry was scored independently and blindly by two investigators (GR, OP), as reported previously [34]. Briefly, Paladin, Ki67 or cleaved-caspase-3 scorings were performed by evaluating extent (%) of each intensity score (ranging from 0 to 3). A global score was the rounded value of the sum of each intensity pondered by its respective extent.
Cell proliferation assay
Cell proliferation was monitored in phase contrast using the Incucyte S3 (Sartorius, Ann Arbor, MI, USA). Cells were seeded in 96-well plates at 10,000 cells per well and images were acquired every 8 h for 96 h. Quantification was performed using the Incucyte software (2021B gui, Sartorius). Each experiment was performed as n = 3 biological replicates.
Wound-healing assay
Wound-healing assays were performed as reported previously [11] using Incucyte S3 (Sartorius, Ann Arbor, MI, USA). Briefly, cells were seeded in 96-well plates at 20,000 cells per well 24 h before the beginning of the experiment to achieve full confluency. Scratches were performed using the Incucyte WoundMaker tool (Sartorius). Wells were imaged every 2 h for 24 h. Quantification was performed using the Incucyte software (2021B gui, Sartorius). Each experiment was performed as n = 3 biological replicates.
Boyden’s chamber chemotaxis migration assay
Cells were suspended in serum-free Dulbecco’s modified Eagle medium (DMEM) supplemented with 0.1% bovine serum albumin (BSA) and 1% penicillin/streptomycin, and seeded (100,000 cells/well) in Boyden Chamber inserts (Costar, 8 µm pore size). The chemo-attractant medium was supplemented with 10% FBS. After 24 h migration, Boyden chambers were cleaned with coton-swaps and then stained with crystal violet. The number of migrating cells was quantified by ImageJ [35]. Each experiment was performed as n = 3 biological replicates.
Co-immunoprecipitation
Total proteins were extracted using a non-denaturing buffer containing Tris-HCl pH 8 (20 mM), NaCl (137 mM), NP40 (1%), EDTA (2 mM) and supplemented with protease inhibitors. Co-immunoprecipitation was performed as described previously [36]. Each experiment was performed as n = 3 biological replicates.
Western blotting
Cells were lysed using 1% sodium dodecyl sulphate with protease and phosphatase inhibitors. Protein separation was then performed using polyacrylamide gel electrophoresis as described previously [37]. Each experiment was performed as n = 3 biological replicates. western blots were quantified using the ImageJ gel analysis tool [35]. Relative quantification was performed according to HSC70 as a loading control.
Subcutaneous injection
Colon cancer cells (106 cells) were suspended in 100 µl DMEM and injected into the flank of 5-week-old NOD-SCID mice randomly assigned to experimental groups. Tumour size was measured weekly during 3–5 weeks. After sacrifice, tumours were resected and measured. Each experiment was performed as n = 3 biological replicates. The sample size was chosen thanks to an a priori T test power evaluation (software G-Power 3.1) [38], using a 40% tumour volume reduction and 95% power.
Intrasplenic injection
Luciferase-positive colon cancer cells (HCT116, LS174T or LoVo—106 cells) were injected into the spleen of NOD-SCID mice, as reported previously [11]. For the experimental metastasis model, splenectomy was performed immediately after cell injection. Mice were imaged each week to assess metastasis spreading using IVIS optical imaging system (Perkin Elmer, Waltham, MA, USA). After 4–6 weeks, mice were sacrificed and imaged.
Establishment of cell clones with differential liver-tropic migratory potential
Invaded spleen and liver collected from the intrasplenic injection in vivo model were dissociated with a mix of collagenase (1600 mg/mL) and hyaluronidase (300 mg/mL) in Hank’s Balanced Salt Solution at 37 °C for 30 min under agitation. The dissociated tissues were then filtrated (70 µm mesh) and cells were seeded in T25 flasks with DMEM supplemented with 10% FBS, 1% penicillin–streptomycin and 0.4% fungizone. Cells were then kept for 4 passages in culture to eliminate fibroblasts. Subsequent cell culture was used to establish cell clones with different migratory potential.
Proteomics and phosphoproteomics
Cells were scraped and lysed in ice-cold lysis buffer (100 mM Tris pH 8.5, 4% sodium deoxycholate). The suspension was thoroughly heated to 95 °C for 5 min before being probe-sonicated three times for 15 sec. Clear lysates were obtained by centrifugation at 18,000 × g for 5 min. Protein concentration was determined by bicinchoninic acid according to the manufacturer’s instructions (Thermofisher Scientific). Samples (500 µg) were diluted with lysis buffer to a final volume of 270 µL before being processed as described previously [39]. At the end of the process, enriched phosphopeptides were reconstituted in 8 µL of solvent A (0.1% trifluoracetic acid in 2% acetonitrile), directly loaded onto reversed-phase pre-column (Acclaim PepMap 100, Thermo Scientific) and eluted in backflush mode. An aliquot (800 ng) of the original peptide mixture was also analysed for total proteome determination. Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC, 0.075 × 250 mm, Thermo Scientific) with a linear gradient of 4–27.5% solvent B (0.1% formic acid in 80% acetonitrile) for 100 min, 27.5%-40% solvent B for 10 min, 40–95% solvent B for 1 min and holding at 95% for the last 10 min at a constant flow rate of 300 nL/min on an Ultimate 3000 RSLC system. Peptides were analysed by an Orbitrap Fusion Lumos tribrid mass spectrometer (ThermoFisher Scientific). The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) coupled online to the nano-LC. Intact peptides were detected in the Orbitrap at a resolution of 120,000. Peptides were selected for MS/MS using HCD setting at 30, ion fragments were detected in the Orbitrap at a resolution of 60,000. A data-dependent procedure that alternated between one MS scan followed by MS/MS scans was applied for 3 sec for ions above a threshold ion count of 5.0E4 in the MS survey scan with 30.0 s dynamic exclusion. The electrospray voltage applied was 2.1 kV. MS1 spectra were obtained with an AGC target of 4E5 ions and a maximum injection time of 50 ms, and MS2 spectra were acquired with an AGC target of 1E5 ions and a maximum injection set to 110 ms. For MS scans, the m/z scan range was 325 to 1800. The resulting MS/MS data were processed using Sequest HT search engine within Proteome Discoverer 2.4 SP1 against a Human database protein database obtained from Uniprot. Trypsin was specified as a cleavage enzyme allowing up to 2 missed cleavages, 4 modifications per peptide and up to 5 charges. A mass error was set to 10 ppm for precursor ions and 0.1 Da for fragment ions. Oxidation on Met (+15.995 Da), phosphorylation on Ser, Thr and Tyr (+79.966 Da), conversion of Gln (−17.027 Da) or Glu (−18.011 Da) to pyro-Glu at the peptide N-term were considered as variable modifications. False discovery rate was assessed using Percolator and thresholds for protein, peptide and modification site were specified at 1%. For abundance comparison, abundance ratios were calculated by Label-Free Quantification (LFQ) of the precursor intensities within Proteome Discoverer 2.4 SP1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [40] partner repository with the dataset identifier PXD030803 and 10.6019/PXD030803. The experiment was performed as n = 3 biological replicates.
Ethics
All animal experimental procedures were performed according to the Federation of European Laboratory Animal Sciences Associations (FELASA) and were accepted after review by the Institutional Animal Care and Ethics Committee of the University of Liège, Belgium (#19-2156). Animals were housed in the animal facility of the GIGA institute.
Statistics
All results are shown as means with a standard error of the mean (sem). Two-sided statistical tests were used according to experimental design. Specific statistical methods are cited in the figures and were chosen according to group number and homoscedasticity. P value < 0.05 was considered statistically significant. Kinase and phosphatase network was constructed by literature mining with Pathway Studio® (May 2020, Elsevier, Amsterdam, The Netherlands). Heatmaps and volcano plots were designed using R v4 installed with pheatmap 1.0.12 and EnhancedVolcano v1.6.0 packages. Gene ontology analysis was performed in Cytoscape v3.9 [41].



留言 (0)