
記住我










Single-cell ribonucleic acid sequencing is becoming widely employed to study biological processes at a novel resolution depth. The ability to analyse transcriptomes of multiple heterogeneous cell types in parallel is especially valuable for cell-focused lung research where a variety of resident and recruited cells are essential for maintaining organ functionality. We compared the single-cell transcriptomes from publicly available and unpublished datasets of the lungs in six different species: human (Homo sapiens), African green monkey (Chlorocebus sabaeus), pig (Sus domesticus), hamster (Mesocricetus auratus), rat (Rattus norvegicus) and mouse (Mus musculus) by employing RNA velocity and intercellular communication based on ligand–receptor co-expression, among other techniques. Specifically, we demonstrated a workflow for interspecies data integration, applied a single unified gene nomenclature, performed cell-specific clustering and identified marker genes for each species. Overall, integrative approaches combining newly sequenced as well as publicly available datasets could help identify species-specific transcriptomic signatures in both healthy and diseased lung tissue and select appropriate models for future respiratory research.
AbstractThe COVID-19 pandemic led to an increase in publicly available single-cell RNA sequencing data. This review provides an up-to-date framework and readily adoptable tools to measure such data in lungs and compare it with existing data across species. https://bit.ly/3wHCoHe
IntroductionSingle-cell RNA sequencing (scRNA-seq) is becoming widely employed to study biological processes in great depth [1–3]. Its name describes its key advantage: the resolution of transcriptomes at single-cell level. This resolution is highly valuable when sample sources are heterogeneous in their cellular compositions, and during the rapid temporal developments occurring during responses to acute infection, such as the increasing cellular intervariability. Thus, scRNA-seq is an essential tool to identify interactive networks, cell fate, disease signatures and novel cell types.
Initially, scRNA-seq was primarily utilised in studies involving oocytes, zygotes and other early developmental cells [4, 5]. Later, global initiatives such as the Human Cell Atlas and CZ Biohub provided the murine [6] and human [7, 8] scRNA-seq atlases. In cell biology focused research, scRNA-seq enabled the discovery of formerly unknown pulmonary cells such as ionocytes [9], aerocytes [10] and group 2 innate lymphoid cells subpopulations [11]. Furthermore, the identification of disease states in individual pulmonary fibrosis derived cells led to the human pulmonary fibrosis atlas, serving as a groundwork for personalised therapies [12]. scRNA-seq was effectively adopted in cancer research, aiming to identify individualised treatment-targets and improve patient care [13–15]. Recently, the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) pandemic further boosted applications of scRNA-seq in lungs due to the need to understand cellular pathomechanisms underlying coronavirus disease 2019 (COVID-19). Here, scRNA-seq became the method of choice to dissect the antiviral immune response both in patient samples and model organisms including transgenic mice, hamsters, ferrets and nonhuman primates [16–21]. Several possible markers predicting severity of COVID-19 cases [22] and potential drug targets [23] were also identified.
From this perspective, pandemic-driven accumulation of substantial lung-centred data allows for the comparison of pulmonary immunity across species and the definition of the advantages and shortcomings of animal models. This requires digital platforms and tools to integrate and compare data derived from human and nonhuman subjects to further support translational research towards new forms of medical diagnosis and (personalised) treatment.
Our main aim is to provide guidance and demonstrate technical feasibility of currently utilised powerful methods for pulmonologists interested in the single-cell transcriptome of lung tissue across different species. For this, we demonstrate current procedures and tools to create, compare and qualitatively assess scRNA-seq data in six different species: human (Homo sapiens), African green monkey (Chlorocebus sabaeus), pig (Sus domesticus), hamster (Mesocricetus auratus), rat (Rattus norvegicus) and mouse (Mus musculus). We show similarities regarding cell differentiation and identify a prominent role of proliferating alveolar macrophages in cell–cell communication.
Basic principles of pulmonary single-cell isolation, barcoding and sequencingLungs harbour a variety of resident and recruited cells, which participate in immune responses and are essential for maintaining organ functionality. Ideally, analyses of lung cells should be performed in full complexity to improve the understanding of various pulmonary pathologies. However, subsequent cross-species cell isolation protocols are rarely identical. Therefore, their potential impact on cellular representation and fitness should be taken into consideration. However, different protocols do not hinder cross-species comparisons, as demonstrated here for selected species and protocol examples (protocols corresponding to datasets depicted in table 1).
TABLE 1Overview of experimental protocols applied to generate viable pulmonary single-cell suspensions; cell isolation procedures corresponding to the interspecies datasets to dissociate lung tissue into a single-cell suspension. Enzymatic digestion and mechanical processing steps applied to dissociate lung tissue into a single-cell suspension vary between species.
Classical droplet-based scRNA-seq technologies such as Drop-seq, In-Drop, Chromium (10× Genomics, Pleasanton, CA, USA) and Nadia (Dolomite Bio, Royston, UK) are widely used due to their high throughput and reasonable pricing. Although recent approaches in bulk RNA and scRNA-seq applications of cryopreserved cells showed promising results for lung epithelial cells, using viable cells from fresh tissue remains the recommended option by the manufacturers (10× Genomics, Dolomite Bio) of commercially available droplet-based scRNA-seq workflows [26, 27]. In plate-based scRNA-seq technologies such as SMART-seq2, single cells are conventionally sorted by flow cytometry into wells in order to overcome the viability shortcoming, as dead cells can be removed during sorting. Another advantage of the SMART-seq2 is the deeper transcriptomic information that can be drawn, much like other full-length RNA-seq methods [28]. The disadvantages include the low number of cells that can be analysed (96 or 384 cells per plate, per run) and significantly higher costs per cell, making it very challenging to utilise for whole organs, especially those that are heterogeneous in structure such as the lungs. The BD Rhapsody system enables high-throughput analyses of the whole transcriptome and adopts a microwell-based approach, resulting in fewer doublet cells compared to the droplet-based 10× Genomics system, albeit with the drawback of allowing the analysis of just one sample per assay [29, 30], an issue that could be solved by additional antibody tagging (cell-hashing).
Ideally, the single-cell suspension, regardless of the method chosen, represents all pulmonary cells in their respective proportion and all original gene signatures. Evidently, cellular dissociation of solid tissues preferably selects migratory cells, such as leukocytes [31]. To dissociate pulmonary mesenchymal, endothelial and epithelial cells, use of enzymatic digestion and enforcement of mechanical stress is necessary [32]. However, these steps increase risks of inducing transcriptomic stress responses that overwrite the cells’ original transcriptomic signature [33–35]. A preventive measure is to add reagents during isolation steps to stop de novo transcription, such as dithio-bis(succinimidyl propionate) [36], CellCover reagent [37] or Actinomycin D [38]. Alternatively, cold-active proteases can be used at low temperatures to hinder ongoing transcription [33]. Another option is single-nucleus RNA sequencing (snRNA-seq), whereby nuclei, rather than viable cells, are isolated from tissues. Advantages of snRNA-seq include the well-established applicability to frozen tissue, minimal isolation-induced cellular stress and improved representation of tissue cells, albeit at the cost of diminished leukocyte frequencies [39, 40]. Particularly, the enhanced representation of pulmonary epithelial and endothelial cells, without need to undergo prior single-cell dissociation steps, makes this technique highly relevant to lung research focusing on tissue responses, e.g. lung barrier integrity in acute lung injury models. Furthermore, snRNA-seq enables the generation of large-scale reference maps from archived tissues as demonstrated by the GTEx Consortium [41]. In autopsy samples from lung, heart, liver and kidney tissues, snRNA-seq outperformed scRNA-seq to systematically access the cellular ecosystem of the tissues [42]. A recent publication comparing scRNA-seq and snRNA-seq data of fresh and cryopreserved murine lung tissue could demonstrate comparable detection rate of genes as well as lower expression of mitochondrial and ribosomal genes, but increased ambient RNA contamination which could not be corrected with the soupX package [40].
Ready-to-use protocols for both human and murine lung tissues are made publicly available, specialising on the isolation of cell nuclei from healthy or diseased (e.g. tumorous) tissues [40, 43, 44].
Once a single-cell suspension has been obtained, the individual cell's RNA can be barcoded, reverse-transcribed and cDNA libraries suitable for next-generation sequencing can be created and sequenced. Each cell's individual barcode enables subsequent reassignment of identified RNA sequences to its cell of origin. For basic analysis allowing for identification of individual cell populations and their specific gene expression, usually 50 000 raw reads per cell are recommended when counting unique molecular identifiers (UMIs), i.e. the number of sequences before PCR-amplification. Enhanced resolution and correct identification of rare transcripts can be achieved through deeper coverage in RNA-seq [45].
Data processing and analysisBioinformatic tools and workflows for processing transcriptomic single-cell data are steadily improving in their scope and usability [46, 47]. The first step in the analysis of each scRNA-seq dataset involves the creation of a table reporting the counts of all detected genes for each measured cell. In the 10× Genomics Chromium system, the vendor's Cell Ranger software is often used. For human and mouse data, the necessary annotation files are publicly available, e.g. by the Encyclopedia of DNA Elements (ENCODE) project [48, 49]; however, it might be necessary for these to be generated from sequencing data for other species. For this, gene annotation of related species can be used to identify genes that also match in the de novo sequenced data as described previously [50]. In the following steps, software tools written in R and Python dominate the field. In R, Seurat [51] and specific packages from Bioconductor [52] are pre-eminent, while in Python, SCANPY [53] is used frequently. Initial filtering of low-quality data is crucial for high-quality results. Such filters ensure a minimum number of unique measured genes and gene counts per cell and remove cells with high percentages of mitochondrial genes, which is an indication of defective cells [46]. Identified doublets (droplets that have erroneously captured more than one cell) are also often removed.
Exemplary workflow for multispecies data pre-processing and integrationIn the following, we demonstrate integration and qualitative assessment of scRNA-seq data across six species from eight datasets: human (Homo sapiens), African green monkey (Chlorocebus sabaeus), pig (Sus domesticus), hamster (Mesocricetus auratus), rat (Rattus norvegicus) and mouse (Mus musculus). We used quality-filtered scRNA-seq data of lungs from different sources (figure 1) using up-to-date analysis workflows. Six datasets are publicly available: resected lung tissue from one patient processed by 1) 10× Genomics (human, Travaglini et al. [24], patient 2) and 2) SMART-seq2 workflow (human, Travaglini et al. [24], patient 2); 3) three hamster lungs (naïve controls from [18]); 4) two rat lungs (data from [25]); 5) two pig lungs (data from [25]); and 6) two monkey lungs (mock-infected controls from [21]). Two further datasets were generated in-house and made available with this article: 7) resected patient lung tissue of four patients (human Charité) and 8) two mouse lungs pooled prior to barcoding (https://github.com/GenStatLeipzig/pulmonologists_interspecies_scRNA). All tissue dissociation procedures for the aforementioned datasets are summarised in table 1.
 FIGURE 1
FIGURE 1 Integration and bioinformatic pre-processing of sequenced lung cells. Schematic depicts single-cell RNA sequencing workflow, created with Biorender.com. FASTQ: text-based format for storing sequence and corresponding quality data; BAM: binary alignment map file format; TSV: tab-separated value file format. #: may require a first round of processing for preliminary clustering; ¶: not for Smart Seq2 data.
As described in the first landmark paper demonstrating cross-species comparison within pulmonary cell types [25], a single unified gene nomenclature is required for subsequent steps. For this, we identified the 9924 human genes with orthologues present in all species from the Ensembl database human genes build GRCh38.p13 (http://www.ensembl.org; supplementary figure S1). In the instance of multiple genes relating to a single orthologue, a consensus gene was selected based on the highest cell-specific expression level. In the following, we refer to the orthologue genes by using the human gene name.
Here, the datasets were similarly transformed by defining common cut-offs for high quality cells, performing regression-based adjustment of the percentage of mitochondrial transcripts (if available), excluding doublets via R-package DoubletFinder [54] and removing ambient RNA via R-package DecontX [55] (figure 1). For the SMART-seq2 dataset, the latter two steps are not required, and a higher cut-off for the minimal number of counts applies as counts here do not reflect UMIs. For normalisation, data was split according to each individual batch and processed with the SCTransform workflow of Seurat making the data more comparable across experiments and species. The SCTransform() function was introduced in 2019 as an improved means of normalisation of scRNA-seq data, based on regularised negative binomial regression [56] and described for integration across different technologies. Applying SCTransform replaces three functions of the previous Seurat workflow: NormalizeData(), ScaleData() and FindVariableFeatures(), we used the version implemented in Seurat_4.0.5. The combining of datasets to create an inter-species data file was performed using Seurat's merge() function, thereby restricting all datasets to genes present in each species. Next, cell-specific clusters with similar expression patterns should be identified. Here, dimension reduction was performed by principal components analysis (PCA), a dimensionality reduction technique to emphasise strong patterns in a dataset considering variation and similarity. R-package Harmony was employed to project the PCA data into a shared embedding [57] to successfully complete the data integration process. The algorithm directs cells to group by cell type rather than according to dataset-specific conditions. As one of many integration tools for scRNA-seq data, Harmony is particularly convincing in terms of usability and was also ranked among top-scoring method [58, 59]. While other top-scoring integration tools are Python-based (scANVI, Scanorama), a second R-based option would be FastMNN or the Seurat IntegrateData() approach applying dimensional reduction to find anchors via reciprocal PCA [59]. However, the method of choice may differ depending on the dataset to be analysed, as exemplified by the systematic comparison in [59]. A comparison between Harmony and Seurat's 4.0.5 function IntegrateData() applying reciprocal PCA is shown in supplementary figure S5, quantification of the quality of integration can be done with measures like integration Local Inverse Simpson's Index (LISI) or cell-type LISI [57].
To visualise the resulting cell types, while taking into consideration the multidimensional space in a clustered two-dimensional plot, uniform manifold approximation and projection (UMAP) was used. A key advantage of UMAP is its ability to map cells according to global differences compared to the previous method of choice (t-distributed stochastic neighbour embedding), which only reflects local differences. Furthermore, it is fast and can be scalable to large numbers of cells. Finally, the number of desired clusters was set by tuning the resolution parameter in Louvain clustering (a graph-based method for community detection that aims to partition a network into clusters of strongly correlated cells) while cells belonging to different clusters were weakly correlated [60]. Advantages of Louvain clustering are speed and scalability as well as the lack of necessity to pre-define the number of clusters [61]. In our analysis, we set the resolution parameter to 0.3, resulting in 22 clusters shared among species. These clusters representing accumulation of cells with similar transcriptomic profiles were annotated to a certain cell type, based on known marker genes and cell-specific annotations transferred from the original dataset. Cell frequencies of both haematopoietic and structural lung cells differ between compared species. While certain differences between species have been described before [62], cell isolation protocols, especially using different enzymatic cocktails, can have a major impact on retrieved cell types. Interspecies research aiming to resolve cell quantitative in addition to qualitative questions should ideally incorporate spatial transcriptomics analyses of tissue sections (figure 2a, supplementary figure S2).
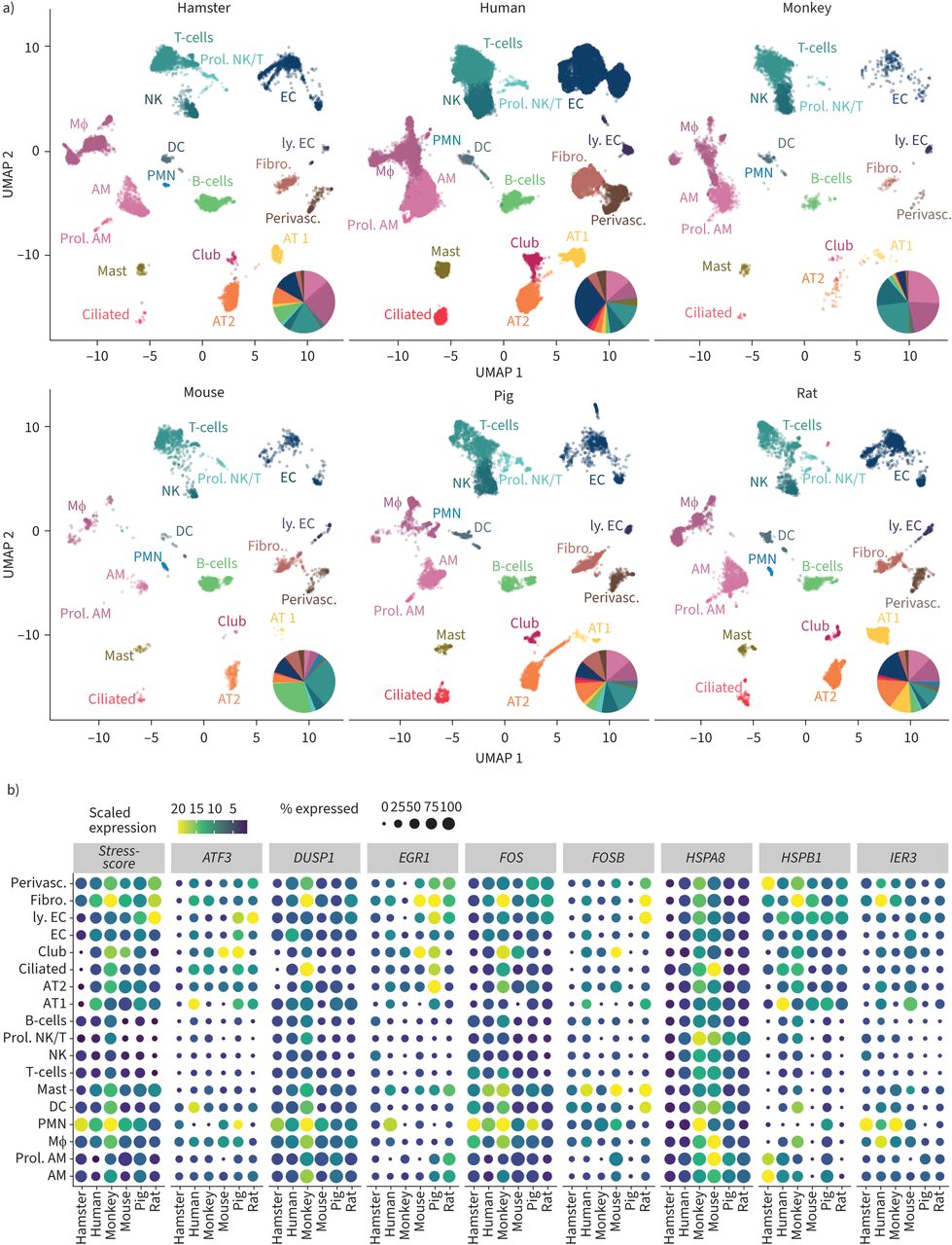 FIGURE 2
FIGURE 2 Cross-species comparison of sequenced lung cells. a) Uniform manifold approximation and projection (UMAP) plot of identified cell populations across species including all datasets, with pie charts of lung cell frequencies in indicated species estimated from single-cell ribonucleic acid sequencing (scRNA-seq) data; b) dot plots indicating the stress response of lung cells across species for all unique molecular identifier based datasets using eight stress-related genes: HSPA8, FOS, DUSP1, IER3, EGR1, FOSB, HSPB1 and ATF3. AM: alveolar macrophages; prol. AM: proliferating alveolar macrophages; Mɸ: interstitial macrophages/monocytes; PMN: polymorphonuclear leukocytes; DC: dendritic cells; mast: mast cells; NK: natural killer cells; prol. NK/T: proliferating NK- and T-cells; AT1: alveolar epithelial cells type 1; AT2: alveolar epithelial cells type 2; ciliated: ciliated cells; club: club cells; EC: endothelial cells; ly. EC: lymphatic endothelial cells; fibro: fibroblasts; perivasc: perivascular cells. Samples: human Charité, cells from four fresh lung explants; human Travaglini et al. [24], data of patient 2; mouse, cells of two whole lungs pooled prior analysis; hamster, cells from lobus caudalis of three hamsters, data from [18]; monkey cells from two lungs, data from [21]; rat and pig, cells from two lungs each, data from [25]. All experiments involving animals were approved by institutional and governmental authorities (Charité Universitätsmedizin Berlin or Freie Universität Berlin and LaGeSo Landesamt für Gesundheit und Soziales Berlin, Germany). Human Charité dataset biomaterial was obtained following approval of the Charité ethics commission, application ID: EA2/079/13, while written informed consent was obtained from all patients.
Cross-species analysis of human and nonhuman scRNA-seq dataTo demonstrate the quality and comparability of data across species, we investigated the expression of eight reputed stress-related genes [34] to monitor putative cell dissociation-associated stress responses. A stress score was developed by averaging cell-wise expression across these genes (figure 2b). Across all analysed species, the applied cell isolation and analysis strategy led to a suitable stress gene signature, with somewhat higher levels for in human lung-derived cells.
As raw sequencing data files were required, we computed RNA velocity (figure 3a, supplementary figure S3) for the in-house generated datasets only [63]. Here, cell development dynamics and temporal expression information were recovered by comparing spliced and unspliced levels of mRNA with an estimated steady-state. The arrows in the two-dimensional UMAP latent space depict an inference of most probable cell state transitions and cell fates based on the transcriptome. The arrow size indicates the coherence and speed of cell development processes in the corresponding region of the UMAP latent space. For velocity estimation, the Python package scVelo was applied with default stochastic model for transcriptional dynamics [64]. By graphical inspection, the most coherent cell movement across the analysed species can be observed in endothelial and T-cell clusters. Interestingly, for human clusters of macrophages and alveolar macrophages, the derived velocities indicate a common area in the UMAP latent space to which the corresponding cells seem to develop. As RNA velocity infers the most probable cell fate solely based on the transcriptome, the compatibility with biological foundations should be verified.
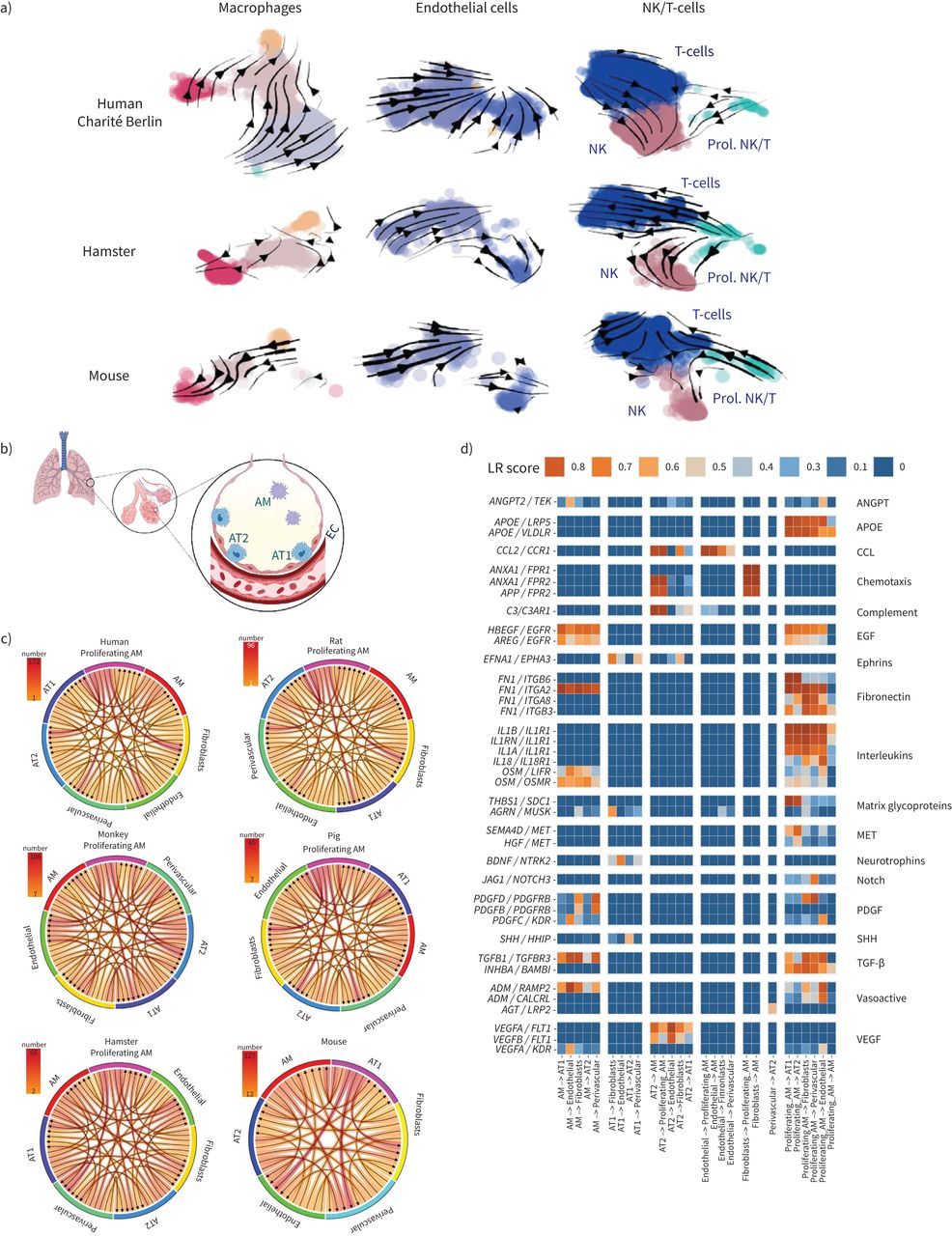 FIGURE 3
FIGURE 3 RNA velocity in pulmonary cells and alveolar niche communication. a) Uniform manifold approximation and projection (UMAP) plots depicting human, hamster and mouse lung cells overlaid with stream arrows derived from the RNA velocity analysis. The colouration indicates chosen cell clusters. b–d) Intercellular communication estimated through ligand–receptor co-expression in the indicated cell types: b) scheme of the healthy alveolus, created with Biorender.com; c) count of relevant receptor–ligand interaction (LRscore ≥0.5) (indicated by arrows) in all species for all unique molecular identifier based datasets. In humans and several animals, proliferating alveolar macrophages appear to show increased intercellular communication. d) Human ligand–receptor pairs found to be conserved (detected in at least one additional nonhuman species) based on the classification of Raredon et al. [25]. Ligand–receptor pairs found only in humans are shown in supplementary figure S4. NK: natural killer cells; prol. NK/T: proliferating NK and T-cells; AM: alveolar macrophages; AT1: alveolar epithelial cells type 1; AT2: alveolar epithelial cells type 2; EC: endothelial cells; ANGPT: angiopoietin; APOE: apolipoprotein E; CCL: C-C motif chemokine ligand; EGF: epidermal growth factor; PDGF: platelet-derived growth factor; SHH: Sonic Hedgehog; TGF: transforming growth factor; VEGF: vascular endothelial growth factor.
Intercellular communication based on ligand–receptor co-expression was calculated based on 3251 recently published pairs available through package SingleCellSignalR [25]. We limited cell-communication analysis to UMI-based datasets available for all species. Through analysed species, proliferating alveolar macrophages seem to dominate ligand receptor interaction within the alveolar spaces (figure 3c and d, supplementary figure S4). All cross-species analysis codes and data, including expression analysis of canonical cell type-specific genes, was made available via Github (https://github.com/GenStatLeipzig/pulmonologists_interspecies_scRNA).
A major limitation is the poor genome annotation for species other than Mus musculus and Homo sapiens, exemplified by that of Mesocricetus auratus (MesAur1.0), which lacks orthologues in ∼25% of all human genes. Integration of further tools into cross-species comparisons, including spatial transcriptomics [65] and new packages from the constantly growing pool of software applications designed for scRNA-Seq data, will further improve our understanding of similarities and differences between cell states and cellular subtypes across species.
OutlookThe meteoric rise of available infection-related patient- and animal model-derived scRNA-seq datasets in response to the COVID-19 pandemic allows for in-depth cross-species comparisons of systemic and pulmonary transcriptomic responses to acute infection. Moreover, such comparisons bare the potential to systematically identify opportunities and limitations of current animal models. Exploiting this unravelled treasure will allow respiratory researchers and pulmonologists to identify universal and species-specific transcriptional responses to pathogens including SARS-CoV-2 and others.
Supplementary materialSupplementary MaterialPlease note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERR-0056-2022.SUPPLEMENT
AcknowledgementsThe authors would like to thank Ulrike Behrendt (Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Infectious Diseases and Respiratory Medicine, Berlin, Germany) for excellent technical assistance. We acknowledge financial support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin and the German Research Foundation (DFG).
FootnotesProvenance: Submitted article, peer reviewed.
Conflict of interest: H. Kirsten reports support for the present manuscript from German Federal Ministry of Education and Research (BMBF) grants e:Med CAPSyS (01ZX1304A) and e:Med SYMPATH (01ZX1906B). E. Wyler has received payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Podcast Gegenblende by DGB, outside the submitted work. M. Landthaler reports support for the present manuscript from Berlin Institute of Health. M. Scholz has received grants or contracts from Pfizer Inc. for a project not related to this research. W.M. Kuebler reports support for the present manuscript from German Research Foundation (KU 1218/9-1, KU 1218/11-1, CRC TR84 A02, CRC TR84 C09, CRC 1449 B01), Germany Ministry for Research and Education (SYMPATH, PROVID consortia), German Center for Cardiovascular Research (Partner site project Berlin) and Berlin Institute of Health (Focus Area Vascular Biology). M. Witzenrath has received grants or contracts from Deutsche Forschungsgemeinschaft, Bundesministerium für Bildung und Forschung, Deutsche Gesellschaft für Pneumologie, European Respiratory Society, Marie Curie Foundation, Else Kröner Fresenius Stiftung, Capnetz Stiftung, International Max Planck Research School, Quark Pharma, Takeda Pharma, Noxxon, Pantherna, Silence Therapeutics, Vaxxilon, Actelion, Bayer Health Care, Biotest and Boehringer Ingelheim, outside the submitted work. M. Witzenrath has received personal fees for consulting from Noxxon, Pantherna, Silence Therapeutics, Vaxxilon, Aptarion, GlaxoSmithKline, Sinoxa and Biotest, and for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from AstraZeneca, Berlin Chemie, Chiesi, Novartis, Teva, Actelion, Boehringer Ingelheim, GlaxoSmithKline, Biotest and Bayer Health Care, outside the submitted work. M. Witzenrath has the following patents planned, issued or pending: EPO 12181535.1: IL-27 for modulation of immune response in acute lung injury (issued: 2012); WO/2010/094491: Means for inhibiting the expression of Ang-2 (issued: 2010); and DE 102020116249.9: Camostat/Niclosamide cotreatment in SARS-CoV-2 infected human lung cells (issued: 2020/21). G. Nouailles receives funding from Biotest AG for a project not related to this work. All other authors have nothing to disclose.
Support statement: G. Nouailles and M. Witzenrath are supported by the German Federal Ministry of Education and Research (BMBF) and the Agence nationale de la recherche (ANR) in the framework of MAPVAP (16GW0247). M. Witzenrath is supported by the German Research Foundation (DFG) grants SFB-TR84 C06, C09, and SFB-1449 B02; the BMBF in the framework of e:Med CAPSyS (01ZX1604B), PROVID (01KI20160A), e:Med SYMPATH (01ZX1906A) and NUM-NAPKON (01KX2021); the BIH (CM-COVID). J. Trimpert is supported by DFG grant number SFB-TR84 Z01b. M-F. Mashreghi receives funding from the State of Berlin and the “European Regional Development Fund” with grant ERDF 2014-2020, EFRE 1.8/11. M-F. Mashreghi is part of the Leibniz Association (Leibniz Collaborative Excellence, TargArt). W.M. Kuebler receives funding from the DFG (SFB-TR84 A02 and C09, SFB 1449 B01, KU1218/9-1 and KU1218/11-1), the BMBF in the framework of e:Med SYMPATH (01ZX1906A), PROVID (01KI20160A), and the German Center for Cardiovascular Research (DZHK) Partner site Berlin. N. Suttorp is supported by DFG (grant number SFB-TR84 B01, C09 and Z02) and BMBF (e:Med CAPSyS grants 01ZX1304B and 01ZX1604B). M. Scholz and H. Kirsten are supported by the BMBF in the framework of e:Med CAPSyS (01ZX1304A), e:Med SYMPATH (01ZX1906B) and V.D. Friedrich within the project “Center for Scalable Data Analytics and Artificial Intelligence (ScaDS.AI) Dresden/Leipzig” (01IS18026B). K. Hoenzke was supported by the SFB-TR84 (DFG), “NUM-COVID 19 Organo-Strat” (BMBF), Charité 3R and the “Einstein Center 3R” (Einstein Foundation Berlin). A.C. Hocke and S. Hippenstiel were supported by Berlin University Alliance GC2 Global Health (Corona Virus Pre-Exploration Project), the SFB-TR84 (DFG), RAPID and “NUM-COVID 19 Organo-Strat” (BMBF) as well as the Berlin Institute of Health (BIH), Charité 3R and the “Einstein Center 3R” (Einstein Foundation Berlin). P. Pennitz and C. Goekeri are supported by Charité 3R. Funding information for this article has been deposited with the Crossref Funder Registry.
Received March 28, 2022.Accepted May 16, 2022.Copyright ©The authors 2022http://creativecommons.org/licenses/by-nc/4.0/This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissionsersnet.org
留言 (0)