
記住我
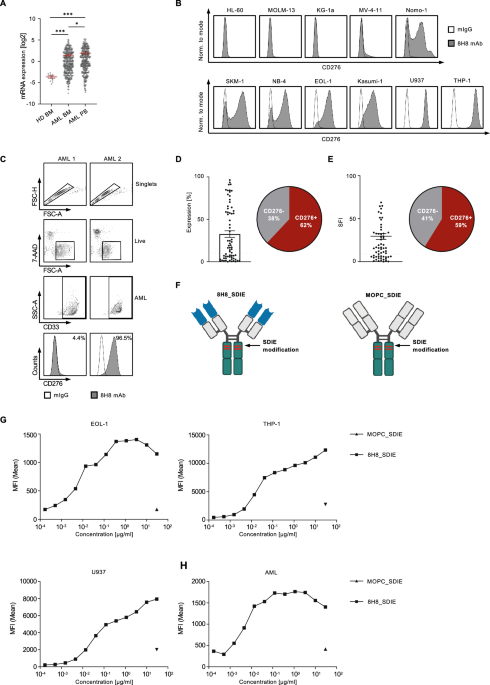
Chimeric antigen receptor T-cell therapy (CAR-T) is a novel class of therapeutics with expanding indications. Currently, CAR-T is FDA approved for relapsed, refractory B-cell acute lymphoblastic leukemia, various subtypes of non-Hodgkin lymphoma (NHL), and multiple myeloma [1].
Therapy-related myeloid neoplasms (t-MN) are aggressive leukemia and associated with high-risk features including complex karyotype (CK), monosomal karyotype (MK), and pathogenic variants (PV) in TP53. Consequentially, the survival following t-MN is generally <1 year. Traditionally, the development of t-MN is associated with exposure to DNA-damaging agents, such as alkylators and radiation, and develops at an average of 5–7 years following the exposure [2, 3]. Recently, exposure to novel modalities such as poly-ADP ribose polymerase (PARP)-inhibitors [4] and peptide receptor radionuclide therapy (PRRT) [5] have been linked to t-MN development.
NHL is one of the most common primary malignancies associated with the risk of a subsequent t-MN. In a series of NHL patients who underwent autologous stem cell transplant (SCT), the 10-year incidence of t-MN was 6% [6]. Older age, having received more lines of therapy, and the use of total body irradiation as a part of the conditioning regimen was associated with a higher risk of t-MN.
The pivotal CAR-T studies in NHL did not report the development of second primary malignancies, including t-MN [7,8,9]. In contrast, recent reports suggested t-MN developing at a median of 3–6 months. However, a systematic study of t-MN following CAR-T and the outcomes thereafter has not been performed. Here, we present the characteristics and outcomes of patients who developed t-MN following CAR-T for NHL and compare outcomes to patients who developed t-MN post SCT.
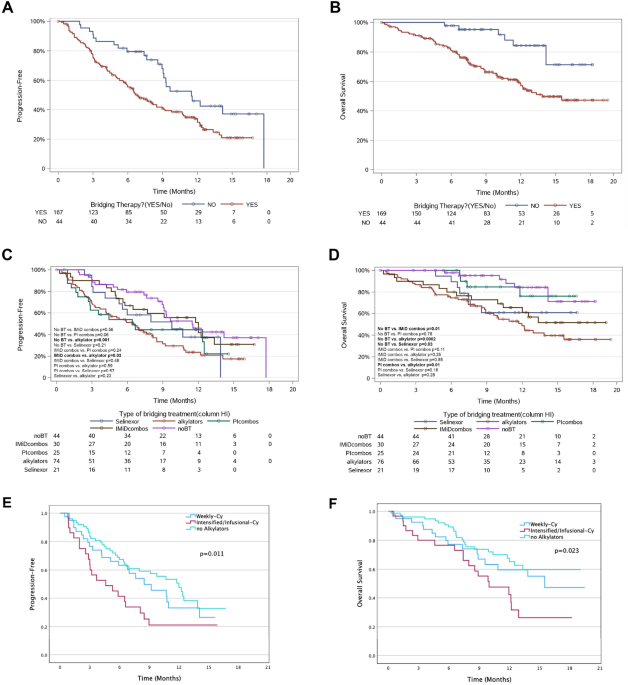
Detailed methods are provided in Supplementary Methods. Between January 1, 2018, and December 31, 2021, 189 patients underwent commercially available CAR-T (93% axicabtagene ciloleucel, 4% brexucabtagene autoleucel, 2% tisagenlecleucel, 1% lisocabtagene maraleucel) for relapsed/refractory NHL at Mayo Clinic. Of these, 10 (5.3%) developed t-MN—five (50%) of whom had received prior autologous SCT, whereas the rest (50%) did not. In those receiving both SCT and CAR-T, the median interval from SCT to CAR-T was 9.8 months (IQR 8.7–23.1). Median interval from CAR-T day 0 to t-MN diagnosis was 9.1 months (IQR 3.6–19.8), with 6 (60%) patients developing t-MN within 1 year from CAR-T. Among the 120 patients with complete follow-up available, the cumulative incidences (CI) of t-MN at 1 and 2 years were 5% and 11%, respectively (Fig. 1A).
Fig. 1: Cumulative incidence and cytopenia associated with therapy-related myeloid neoplasm (t-MN) development in non-Hodgkin Lymphoma patients undergoing chimeric antigen receptor (CAR)-T therapy.
A Cumulative incidence of t-MN in NHL patients undergoing CAR T cell therapy at 2 years. B Complete blood count at day +30 post CAR T-cell therapy, day +100 post-CAR T-cell therapy, and at t-MN diagnosis. Hemoglobin in g/dL, platelets ×109/L, white blood cell count (WBC)—×109/L, and absolute neutrophil count (ANC)—×109/L.
Clinical and laboratory characteristics of patients who developed t-MN following CAR-T are described in Supplementary Table 1. Prior to CAR-T, all patients underwent bone marrow aspirate and biopsy evaluation. At the time of pre-CAR-T bone marrow evaluation, median hemoglobin was 9.9 g/dL (IQR 8.8–10.7), white blood cell (WBC) count was 3.5 × 109/L (IQR 2.5–4.3), absolute neutrophil count (ANC) was 2.2 × 109/L (IQR 1.3–2.9), and platelet count was 115 × 109/L (IQR 61–168). Upon morphology review of pre-CAR-T bone marrow, none were noted to have morphologically diagnostic features of t-MN. We retrospectively performed NGS using the pre-CAR-T bone marrow sample in three patients. All three patients harbored PVs: patient #1251 had DNMT3A (Arg882His, VAF 7%), patient# 1724 had DNMT3A (Met801Val, VAF 8%), and patient# 2035 had TP53 (Ile254Ser, VAF 40%).
All patients received fludarabine and cyclophosphamide lymphodepletion chemotherapy followed by the infusion of axicabtagene ciloleucel. Peak cytokine release syndrome (CRS), neurotoxicity, and response rates at day +30 and +90 are provided in Supplementary Table 1. Median hemoglobin, WBC, ANC, and platelet counts at day +30 were 8.3 (IQR 8–8.7), 2 (IQR 1.7–4.8), 1.3 (IQR 1.1–2.4), 33.5 (17.8–56.8) and at day +100 was 8.55 (IQR 7.7–9), 2.55 (IQR 1.9–3.4), 1.5 (IQR 0.9–2), and 41.5 (IQR 27–78.5), respectively (Fig. 1B).
At t-MN diagnosis, the median age was 66.2 years and 70% were female (Supplementary Table 2). Eight (80%) patients presented with t-MDS, whereas 20% presented as t-AML. CBC parameters at t-MN diagnosis were not different from day +30/+90 assessments: hemoglobin 8.8 (IQR 7.4–9.3), WBC was 2 (IQR 0.9–4.2), ANC was 0.7 (IQR 0.5–1), and platelet count was 21 (IQR 14–37). Nine (90%) patients had a cytogenetic abnormality, of which 4 (40%) had CK/MK. NGS at t-MN diagnosis was available in 9 (90%) patients with TP53 being the most commonly mutated gene (4, 44.4%). Paired genetic analysis of pre-CAR-T and t-MN bone marrows (n = 3) showed that DNMT3A clones detected in the pre-CAR-T bone marrow were not detectable at t-MN in both patients—one (UPIN# 1251) developed RUNX1-mutated t-MN with deletion 20q, and the other (UPIN# 1724) had deletion 13q, but no detectable PVs. UPIN# 2035 was noted to have the expansion of the TP53 clone (VAF 40 to 84%) and CK/MK at t-MN diagnosis.
We next compared t-MN that developed following the two cellular therapy platforms: patients who developed t-MN following SCT (SCT t-MN cohort, n = 28) and CAR T-cell therapy (CAR-T t-MN cohort, n = 10). As noted above, 5 of the 10 CAR-T t-MN patients had received a prior autologous SCT. The clinical and laboratory characteristics of the two cohorts are shown (Supplementary Table 3). CAR-T t-MN cohort patients received more lines of therapy (5 vs. 4, P = 0.02), though the cumulative doses of doxorubicin (262.5 vs. 300 mg/m2, P = 0.025) and melphalan (70 vs. 140 mg/m2, P = 0.01) were lower. The cumulative doses of the other alkylators as well as the proportion of the patients who received platinum-based or nucleoside analog-based chemotherapies were not different. At t-MN diagnosis, CAR-T t-MN patients had lower hemoglobin (6.8 vs. 8.7 g/dL, P = 0.008), ANC (2 vs. 4.6 × 109/L, P = 0.016, and platelet count (21 vs. 65 × 109/L, P = 0.009). Cytogenetic and genetic profiles including the proportions of patients with CK, MK, and PV in TP53 were not different between the two cohorts.
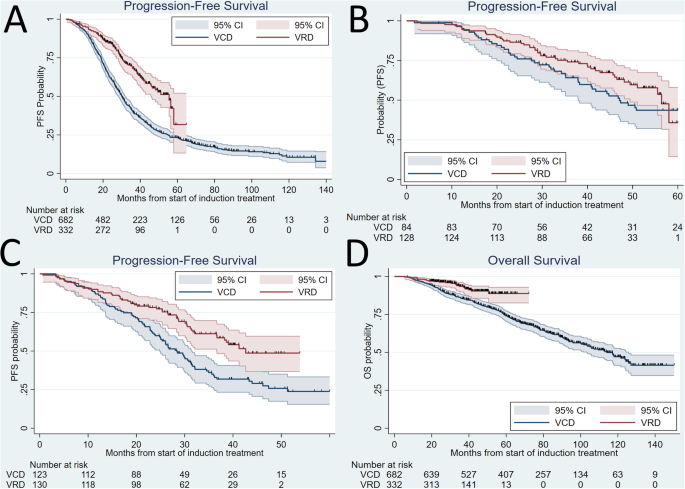
The median myeloid-neoplasm-free survival (MNFS) from the first intervention was shorter for CAR-T t-MN patients compared to SCT t-MN patients (43 vs. 100 months, P = 0.01, Fig. 2A). This observation remained valid when patients who received both SCT and CAR-T (n = 5) were excluded (29 vs. 100 months, P = 0.01, Fig. 2B). Similarly, the median MNFS from day 0 for CAR-T t-MN cohort was shorter compared to the SCT t-MN cohort—both when patients who received both SCT and CAR-T were included (22 vs. 44 months, P = 0.01, Fig. 2C), or excluded (11 vs. 44 months, P < 0.001, Fig. 2D). The median MNFS for CAR-T t-MN was not different when stratified by having received prior SCT (n = 5) or not (n = 5, 27 vs. 11 months, P = 0.11), though this analysis was limited by small sample size. Finally, overall survival following t-MN development was not different between the cohorts (Fig. 2E, F).
Fig. 2: The latency to develop therapy-related myeloid neoplasm is significantly shorter following chimeric antigen receptor (CAR)-T therapy compared to autologous stem cell transplant (SCT) in NHL.
A Myeloid neoplasm-free survival (MNFS) from the first intervention comparing patients who underwent SCT (n = 28) with those who underwent CAR T cell therapy with or without prior SCT (CAR-T, n = 10). B MNFS from the first intervention comparing patients who underwent SCT only (n = 28) or CAR-T only (n = 5). C MNFS from day 0 comparing patients who underwent SCT (n = 28) with those who underwent CAR-T with or without prior SCT (n = 10). D MNFS from day 0 comparing patients who underwent SCT only (n = 28) or CAR-T only (n = 5). E Comparing OS from t-MN diagnosis in patients who underwent SCT (n = 28) with those who underwent CAR-T with or without prior SCT (n = 10), and F comparing OS from t-MN diagnosis in patients who underwent SCT only (n = 28) or CAR-T only (n = 5).
Our two most notable observations include a relatively high incidence of t-MN and the short interval from CAR-T to t-MN. No prior studies have reported the CI of t-MN following CAR-T. On the other hand, short latency appears to be a common theme in post-CAR-T t-MN. For example, Shouse et al. identified four patients who developed t-MN at a median interval of 3 months (range 2–3) [10], and Cordeiro et al. observed 4 of 86 patients developed t-MN at a median of 6 months (range 4–17) [11]. In both cases, a subset of patients was noted to have evidence of dysplasia [10] or clonal abnormalities prior to CAR-T therapy [11].
Potential explanations for the shorter latency and a substantially higher CI of t-MN following CAR-T compared to historical observations therapies [3, 12, 13], include the additional DNA-damaging therapy received, improved survival due to better lymphoma control, or that CAR-T, by yet an unknown mechanism, increased the risk of t-MN. While patients who received CAR-T indeed received a median of one additional line of therapy, cumulative exposures to the known DNA-damaging agent were not different. Second, the interval from day 0 to t-MN was not different between CAR T-cell patients whether they received prior SCT or not. Finally, we consistently observed shorter MNFS in the CAR-T t-MN cohort—both when calculated from the first intervention or day 0. However, proving the causality of t-MN is challenging [12], especially as NHL patients receive multiple lines of DNA-damaging therapies over a variable interval. Moreover, there is a debate that SCT acts as an additional risk factor for t-MN [6, 12]. Given that half of the CAR T-cell t-MN cohort also received prior SCT, this small retrospective analysis does not allow for establishing CAR-T as a contributory factor for t-MN and the mechanism underlying our observations remains undetermined. Other limitations of our study include those of single-institution retrospective studies. Second, it has not been our practice to obtain cytogenetics and NGS on all NHL patients undergoing CAR-T, limiting our ability to track the clonal evolution before and after CAR-T.
Our observations have notable implications. First, given the strikingly short interval between CAR-T and the development of t-MN, all patients undergoing CAR-T for NHL should have a careful discussion regarding this potentially fatal complication. Second, we and others noted that a subset of patients had dysplasia [10] or clonal abnormalities [11, 14] before CAR-T. Therefore, a careful bone marrow evaluation including cytogenetics and NGS may be beneficial [14]. Finally, as the utilization of CAR-T increases; surveillance, diagnosis, and treatment of t-MN should be an essential component of the long-term survivorship plan.
ReferencesJune CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73.
Higgins A, Shah MV. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes. 2020;11:749.
McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:513.
Morice P-M, Leary A, Dolladille C, Chrétien B, Poulain L, González-Martín A, et al. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: a safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021;8:e122–e34.
Sonbol MB, Halfdanarson TR, Hilal T. Assessment of therapy-related myeloid neoplasms in patients with neuroendocrine tumors after peptide receptor radionuclide therapy: a Systematic Review. JAMA Oncol. 2020;6:1086–92.
Radivoyevitch T, Dean RM, Shaw BE, Brazauskas R, Tecca HR, Molenaar RJ, et al. Risk of acute myeloid leukemia and myelodysplastic syndrome after autotransplants for lymphomas and plasma cell myeloma. Leuk Res. 2018;74:130–6.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory Mantle-cell lymphoma. N Engl J Med. 2020;382:1331–42.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2018;380:45–56.
Shouse GP, Xue T, Herrera A, Siddiqi T, Zain J, Popplewell L. et al. MDS as a cause for prolonged hematologic toxicity after treatment with CD19 targeted CAR-T cell therapy in patients with relapsed refractory lymphoma. Hematol Oncol. 2019;37(S2):507–8.
Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol Blood Marrow Transplant. 2020;26:26–33.
Armitage JO, Carbone PP, Connors JM, Levine A, Bennett JM, Kroll S. Treatment-related myelodysplasia and acute leukemia in non-Hodgkin’s lymphoma patients. J Clin Oncol. 2003;21:897–906.
Micallef INM, Lillington DM, Apostolidis J, Amess JAL, Neat M, Matthews J, et al. Therapy-related myelodysplasia and secondary acute myelogenous leukemia after high-dose therapy with autologous hematopoietic progenitor-cell support for lymphoid malignancies. J Clin Oncol. 2000;18:947.
Miller PG, Sperling AS, Brea EJ, Leick MB, Fell GG, Jan M, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 2021;5:2982–6.
AcknowledgementsWe are grateful to our patients and their families. MVS was supported by Paul Calabresi Program in Clinical/Translational Research at Mayo Clinic (K12CA090628); Leukemia Research Foundation New Investigator Award; and the Eagles Foundation.
Author informationAuthors and AffiliationsContributionsHBA and MVS abstracted data and wrote the first draft of the manuscript; HBA, RM, AR, HM, MK-D, JCBV, NB, SMA, MMP, AA-K, SSK, YL, and MVS accrued patients; PG analyzed cytogenetics and acquired the samples for NGS, RH performed and interpreted NGS analysis; RB, MH, and YL provided outcomes data, DC performed pathology review, MVS conceived the project, and performed the statistical analysis. All authors reviewed and edited the manuscript and agree to the submitted version.
Corresponding authorEthics declarations Competing interestsAA-K—Research support to institution: Novartis, Astex. MMP—Membership on an entity’s Board of Directors or advisory committees (Stemline Therapeutics) and Research funding (Kura Oncology). PG—Membership on an entity’s advisory board (AbbVie) Kharfan-Dabaja- Consultancy for Daiichi Sankyo. SSK—Patents and Royalties: Novartis, Humanigen, MustangBio, and Mettaforge; Research funding: Novartis, Kite/Gilead, Juno/BMS, Lentigen, Humanigen, Tolero, Sunesis/Viracta, MorphoSys, and Leahlabs; Scientific Advisory Boards: Novartis, Kite/Gilead, Juno/BMS, Humanigen; DSMB: Humanigen; Consultancy: Novartis, Torque, and Calibr.
Additional informationPublisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary informationRights and permissionsOpen Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article Cite this article
Cite this articleAlkhateeb, H.B., Mohty, R., Greipp, P. et al. Therapy-related myeloid neoplasms following chimeric antigen receptor T-cell therapy for Non-Hodgkin Lymphoma. Blood Can
留言 (0)