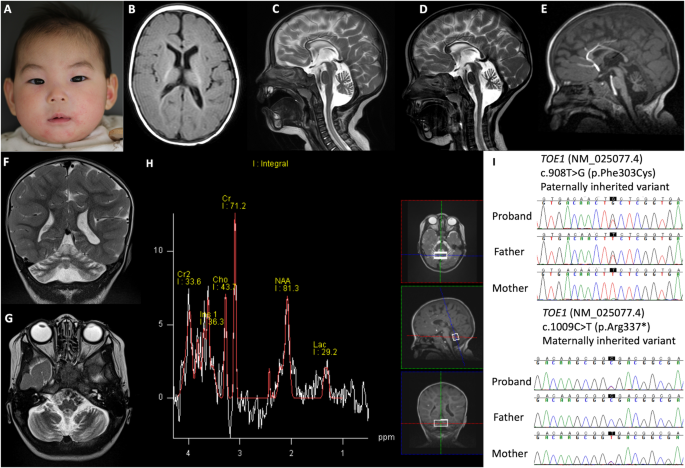
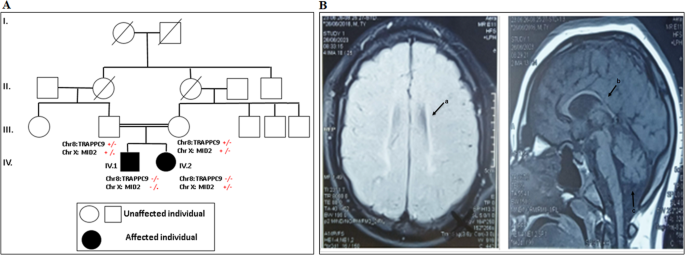
A homozygous loss-of-function variant in BICD2 is associated with lissencephaly and cerebellar hypoplasia
The identified variant p.(Gln77Ter) is new and absent from the Genome Aggregation Database. It was evidenced that pathogenic variants in BICD2 are extremely rare in the population, predicted to be damaging by most tools, and occur in specific hotspots within key BICD2 functional domains [8]. Furthermore, WES did not identify any variant(s) in any of the OMIM genes with an acknowledged disease association (including VPS13B gene). Although BICD2 is essential for the proper development of the cerebral cortex [5] but there have been no other clinical reports of individuals with loss of-function variants in BICD2 showing lissencephaly and cerebellar hypoplasia. However, lissencephaly and cerebellar hypoplasia are consistent with that observed after BICD2 knockdown in mice showing defects in laminar organization of the cerebral cortex, hippocampus and cerebellar cortex, indicative of radial neuronal migration defects. Cell-specific inactivation of BICD2 in astrocytes and neuronal precursors revealed that radial cerebellar granule cell migration is non-cell-autonomous and intrinsic to cerebellar Bergmann glia cells [9, 10]. Therefore, we considered BICD2 to be a convincing candidate gene in the context of lissencephaly and cerebellar hypoplasia. The absence of homozygous loss of function BICD2 variants in the healthy family members supports the clinical relevance of BICD2.
Recently, biallelic variant c.731 T > C p.(Leu244Pro) in BICD2 was described in a girl with abnormal gyral pattern in fronto-temporo-parietal regions [6] (Table 1). The girl displayed additionally moderate intellectual disability and Cohen-like features [6]. In comparison, our patient showed congenital microcephaly, profound delay, seizures, lissencephaly and cerebellar hypoplasia. Unlike the patient with Cohen-like features, our patient showed spasticity and developed contracture deformities and did not show neutropenia. Interestingly, a heterozygous missense variant c.2080 C > T, p.(Arg694Cys) was reported in two unrelated patients with mild perisylvian polymicrogyria, and mild cerebellar vermis hypoplasia [4]. Moreover, a BICD2 nonsense variation p.(Lys775Ter) was identified in a boy with lissencephaly and subcortical band heterotopia [5]. These heterozygous variants are located within the highly conserved CC3 domain of BICD2 (Table 1). Nevertheless, the heterozygous missense variants within the CC1 domain were not associated with abnormalities of cortical development but even showed a milder course of SMALED2A and a higher frequency of foot deformities [8]. Indeed, a larger cohort is required to draw conclusions regarding genotype-phenotype correlations.
Table 1 The clinical findings and variants identified in patients with BICD2 and brain anomaliesLissencephaly and cerebellar hypoplasia noticed in our patient appeared similar to those with LIS1 variants. This is not surprising as LIS1 interacts with the dynein/dynactin complex and BICD2 to recruit cellular structures [11]. In the mean time, these brain MRI features may overlap with RELN-mutated patients phenotype. However, the cortical migration defect was more severe in our patient than in RELN-mutated patients. In addition, our patient had mild cerebellar hypoplasia unlike RELN-mutated patients who had profoundly hypoplastic and dysplasic cerebellum with no identifiable folia [12].
Our study provides valuable findings into human developmental brain malformations disorders associated with definitive loss-of function variants in BICD2.

留言 (0)