
記住我
Ten males exercised intermittently (60 min) on a cycle ergometer (Additional file1: Fig. S1). The pulmonary O2 consumption rate (VO2) and blood lactate concentration immediately after the high-intensity bouts of intermittent exercise were maintained above 80% of maximal VO2 and 5 mM, respectively (Additional file1: Fig. S1) (i.e., the relative intensity of exercise was high, and the exercise induced substantial metabolic stress without progressive metabolite accumulation). Biopsy samples from the m. vastus lateralis were taken prior to and at 2 min, 1 h, 3 h, and 6 h after exercise for CAGE (Additional file1: Fig. S1).
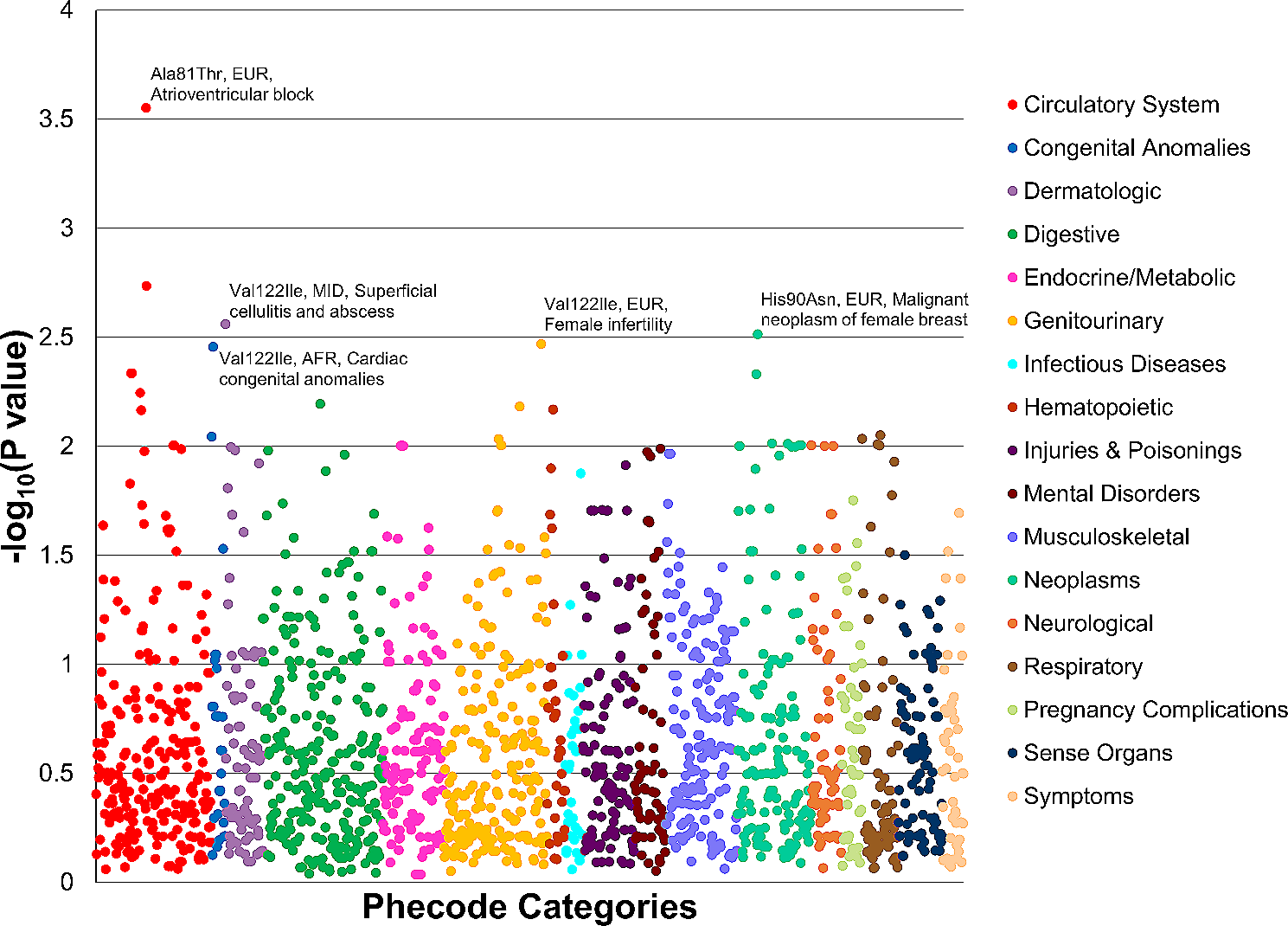
Annotation of CAGE TSSsIn total, we identified 44,680 CAGE-tag-defined TSS clusters (CAGE TSS clusters) (Additional file2: Table S1). In our study, the CAGE TSS clusters showed the classical distribution into “sharp” and “broad” classes of promoters (Fig. 1A), as described previously [3]. To annotate CAGE TSS clusters to genes, we used both the Ensembl and RefSeq annotations. Most of our CAGE TSS clusters fall between ± 50 bp from the annotated TSSs (Fig. 1B). Hence, we used that interval to annotate CAGE TSS clusters as previously annotated TSSs; the annotation priority for other CAGE TSS clusters is shown in Fig. 1C. In total, we annotated 41,951 CAGE TSS clusters to the previously annotated TSSs, exons, and non-coding regions (Additional file2: Table S1). It is noteworthy that elimination of low-abundance CAGE TSS clusters (10% cutoff; see Materials and Methods) substantially reduced (by 30%) the number of annotated CAGE TSS clusters, particularly the number of CAGE TSS clusters annotated to the coding sequence (CDS; by 94%) and 3′ untranslated region (UTR; by 91%) (Fig. 1D; Additional file2: Table S1). These eliminated TSSs belonged predominantly to muscle-specific genes with high expression and/or many exons (e.g., titin, nebulin, and myosin heavy chain 1, 2, and 7) and are probably related to biological and/or technical noise.
Fig. 1
Annotation of cap analysis of gene expression (CAGE) transcription start site (TSS) clusters. A. CAGE TSS clusters showed the classical distribution to “sharp” and “broad” classes. B. Distribution of distance from CAGE TSS clusters to TSSs annotated in Ensembl and RefSeq (most of our CAGE TSS clusters fall between ± 50 bp from annotated TSSs). C. Location of CAGE TSS clusters (ordered by priority) versus gene annotation. Additionally, putative alternative starts were verified using the coverage and exon–exon junction (RNA sequencing data) and annotated to corresponding genes. D. Number of CAGE TSS clusters annotated to different locations before (left panel) and after (right panel) elimination of low-abundance CAGE TSS clusters. E. Expression (median and interquartile range) of CAGE TSS clusters annotated to different locations. F. Overlap of CAGE TSS clusters from the FANTOM5 and refTSS projects with those in our study and 3764 first defined (probably muscle-specific) CAGE TSS clusters. G. Analysis of the coverage and exon–exon junctions for the first exon (RNA sequencing data; example for the NOS1 gene) allows the annotation, for the first time, of 290 CAGE TSS clusters belonging to 163 genes. CDS, coding sequence
The remaining CAGE TSS clusters were compared with the RNA sequencing data from our previous study investigating the effect of aerobic exercise training in human skeletal muscle [15]; verification by coverage and exon–exon junctions for the first exon allowed us to annotate for the first time 290 CAGE TSS clusters belonging to 163 genes (Additional file2: Table S1; Fig. 1D, G). Among them were several well-known protein-coding genes, including nitric oxide synthase 1 (NOS1), calcium/calmodulin-dependent protein kinase II alpha (CAMK2A), E1A binding protein P300 (EP300), ribosomal protein S6 kinase A2 (RPS6KA2), homeodomain interacting protein kinase 2 (HIPK2), angiomotin (AMOT), and homeobox A11 (HOXA11), as well as some pseudogenes and long non-coding RNAs. The remaining 2911 CAGE TSS clusters were annotated to introns and intergenic regions (Fig. 1D). The mean expression level of the CAGE TSS clusters annotated to these locations was very low. In contrast, the CAGE TSS clusters verified by RNA sequencing (n = 290) show high mean expression level, similar to the CAGE TSS clusters annotated to TSSs and 5’UTRs (Fig. 1E). This finding indirectly confirms the biological significance of the CAGE TSS clusters verified by RNA sequencing in our study.
The CAGE TSS clusters defined in our study overlapped to a large extent with those defined in various human tissues and cells in the FANTOM5 and refTSS projects, which analyzed a limited number of human skeletal muscle samples. This partially explains why we found 3764 new (probably muscle-specific) CAGE TSS clusters (Fig. 1F). However, half of these CAGE TSS clusters were annotated to introns, intergenic regions, and genes verified by RNA sequencing data (Fig. 1F). As mentioned above, only a small fraction of these CAGE TSS clusters showed a high expression level (mainly the CAGE TSS clusters verified by RNA sequencing) (Fig. 1F).
Alternative TSSs contribute significantly to stress-induced transcriptome responseAcute exercise changed (mainly upregulated) expression of several hundred genes with a relatively small degree of overlap at each time point (in total 1411 differentially expressed genes [DEGs]) (Fig. 2A, Additional file3: Table S2). Principal component analysis showed that gene responses to exercise at different time points fell into different clusters, confirming the consistency of gene responses in different volunteers (Fig. 2B). Those findings suggest that the transcriptome dynamically changes during the first hours after acute stress. In agreement with previous studies [13, 15], the most highly enriched biological process for upregulated genes in human skeletal muscle was regulation of transcription (Additional file1: Fig. S2, Additional file3: Table S2). This is in line with findings in human and mouse cells showing domination of mRNA TFs in the earliest responses to various stimuli [16].
Fig. 2
Contribution of alternative TSSs to acute-stress-induced transcriptome response and their functional role. A. Number of differentially expressed genes (DEGs) relative to pre-exercise and their overlap at different time points after acute exercise. Up- and down-regulated genes are shown in red and blue, respectively; the number of time-specific DEGs is underlined. B. Principal component analysis shows the consistency of gene responses in different volunteers at different time points during the first hours after acute exercise. C. Number of DEGs having one (532 genes) or several (733 and 146 genes) TSSs and showing differential TSSs usage (146 genes). Another set of genes (111 genes) shows differential TSSs usage without altering the overall expression of each gene. D. Examples of different TSS regulation patterns. E. Distribution of TSSs per gene and a potential functional role of the alternative TSSs
Two-thirds of DEGs have two or more TSSs (alternative CAGE TSS clusters) (Fig. 2C and D), meaning that stress-induced gene expression is related to the regulation of alternative starts. Interestingly, the change in the expression of 146 (~ 10%) of DEGs was associated with the activation of alternative TSSs, indicating differential TSSs usage (Fig. 2C, D, Additional file3: Table S2). Moreover, in another set of 111 genes, differential TSSs usage occurred without altering overall gene expression (the sum of all TSSs related to a gene) and was associated with multidirectional changes in the expression of various TSSs in a gene (Fig. 2C, D, Additional file3: Table S2).
In line with findings in various human tissues [3, 4], we found that 7591 of 12,268 expressed genes have more than one CAGE TSS clusters (Fig. 2E), suggesting that skeletal muscle has a high potential for generating alternative mRNA isoforms. Moreover, 948 of 7591 genes have at least one removed alternative promoter (> 200 bp beyond the promoter region of the canonical TSS—most highly expressed at baseline; see below) of which 89 demonstrate differential TSS usage (Additional file4: Table S3). Importantly, these genes mainly involved in regulation of transcription (Gene Ontology analysis; Additional file3: Table S2), indicating the important contribution of the alternative TSSs belonging to the removed alternative promoters in response to exercise-induced stress. If the alternative TSS is located up- or downstream of the 5′ UTR of the canonical TSS, then this can lead to the appearance of a new first exon(s) and another amino acid residue at the N-terminus (Fig. 2E), which may change the function of the protein. Given the diversity of exon–intron structures of already known mRNA isoforms, predicting all possible mRNA isoforms based on CAGE TSS clusters is a difficult task. Therefore, using data on known mRNA isoforms with defined start codons (Ensembl) and on our CAGE TSS clusters (see Materials and Methods), we found that 197 genes have alternative start codons associated with annotated alternative protein isoforms (Fig. 2E, Additional file4: Table S3). This list can be increased by data from CAGE TSS clusters, which we annotated in our study for the first time (Fig. 1D, G, Additional file2: Table S1). If we assume that most of the alternative mRNA isoforms encoding alternative protein isoforms are already known, then the presence of several TSSs in each of ~ 7000 genes means that the main function of alternative starts is associated not only (to a limited extent) with the generation of alternative protein isoforms but also with the fine regulation of expression of the mRNA isoform from a gene due to the activation of various alternative starts by TFs specific to them (e.g., when the alternative start is located in the 5′ UTR) (Fig. 2E).
Localization of the individual promoter regions surrounding CAGE TSSs in muscleThe positional weight matrix (PWM) method is a classic approach most frequently used for prediction of TFBSs and corresponding TFs responsible for stress-induced DEGs. Estimation of the exact location of each TSS for each gene and the expression level of each TSS is crucial for correct prediction of TFs. Usually, the size of the region around the TSSs in which TFBSs are sought (i.e., a conditional promoter region) is chosen empirically from − 1500– + 500 bp to − 300– + 100 bp (i.e., so-called standard promoter regions). However, the size of promoter regions determined by open chromatin differs significantly for different genes [17, 18]. Therefore, the use of an individual promoter region for each TSS is necessary for the correct prediction of TFs. Because the position of open chromatin can differ between different cells [19, 20], we determined the open chromatin surrounding the CAGE TSS clusters using data from experiments with human gastrocnemius medialis muscle: (see “Methods” section). To identify the individual promoter regions in skeletal muscle, we used the overlap of open chromatin evaluated by ATAC-seq and DNase-seq signals around each CAGE TSS cluster (Fig. 3A). We found an open chromatin probability distribution markedly shifted upstream of the CAGE TSSs (Fig. 3B), as shown previously in yeast [17] and mice macrophages [18].
Fig. 3
Individual promoter regions for the CAGE TSSs in skeletal muscle. A. Individual muscle promoter region around the CAGE TSS cluster was identified using overlapped ATAC-seq and DNase-seq data (MACS2 peaks) (example for a bidirectional promoter); additionally, the density of TFBSs was shown (see Additional file1: Fig. S3). B. Open chromatin probability distribution around the CAGE TSS (− 2000 to + 2000 bp) is similar to the density of transcription factor binding sites (TFBSs) (thick and dashed lines are median and interquartile range, respectively). C. Individual muscle promoters (86%) show greater expression level and density of TFBSs than a small fraction of pseudo-promoter—a region − 2000 to + 2000 bp from the CAGE TSS clusters without (> 2000 bp from the CAGE TSS cluster) open chromatin. Median, interquartile range, 1–99% range, and P value (Wilcoxon test) values are shown. D. The individual muscle promoter regions for all promoters and bidirectional promoters are shown (arranged, from bottom to top, according to increasing total length of the promoter; each bidirectional promoter is depicted twice in relation to each TSS). E. The use of individual promoter regions increases the number of predicted TFs associated with changes in gene expression at 1 h, 3 h, and 6 h after exercise, compared with the use of standard regions with a fixed length
In addition to open chromatin, a promoter region is characterized by a high density of TFBSs for various TFs [17, 18, 21, 22], which means a high potential for DNA in this region to bind with various TFs. Using data from 15,982 chromatin immunoprecipitation (ChIP)-seq experiments with human cells and tissues (the GTRD database [23]), we determined the density of TFBSs on the DNA around each CAGE TSS (− 2000 to + 2000 bp). Figure 3B shows that the density of TFBSs is similar to the open chromatin probability distribution. Importantly, only a small fraction of the CAGE TSS clusters showed no open chromatin near a CAGE TSS clusters (> 200 bp beyond a CAGE TSS clusters). As expected, these TSSs demonstrate substantially lower expression and TFBS density than those with open chromatin (Fig. 3C).
Because the CAGE TSSs for gene(s) may be located near each other on the same or opposite strand (bidirectional promoters), such CAGE TSSs fall to one promoter region (Fig. 3A). These bidirectional promoters constitute 12% of all promoters and show similar density of TFBSs compared with other promoters with open chromatin (Fig. 3C). As a result of the overlap analysis, we distinguished the coordinates for 11,830 promoter regions in muscle (belonging to 90% of the genes defined in our study) (Fig. 3D, Additional file4: Table S3). No difference in the total length distribution was found between bidirectional and other promoters (Fig. 3D). Then, we identified differentially regulated (exercise-induced) promoter regions in muscle; these promoters demonstrate excellent coincidence with exercise-induced DEGs identified in the study by another bioinformatics approach (Additional file4: Table S3, Fig. 2A).
To examine the ability of the individual promoter regions to better predict TFs than the standard promoter regions, we identified TFs associated with exercise-induced (differentially regulated) promoter for several standard and individual promoter regions by the PWM method (using the TRANSFAC Database v.2020.3 [24] containing matrices for 1357 of 1639 known TFs [25]). The number of predicted TFs with strong adjusted fold enrichment values > 1.5 were then compared. Figure 3E shows that if individual muscle promoter regions are used, the number of predicted TFs is substantially greater than that of standard promoter regions.
TFs associated with differentially regulated individual promoter regions in musclePromoters regulated by a set of TFs should demonstrate similar dynamics of gene expression. Using an unsupervised analysis (the Chinese restaurant process), we identified 21 clusters with co-expressed (presumably co-regulated) differentially regulated individual promoter regions. Then, using the PWM method, TFBSs enriched in the individual promoter regions were predicted in each cluster (Fig. 4, Additional file5: Table S4). Figure 4 shows the average ranked expression of the individual promoter regions and the most enriched TFs for each cluster. Importantly, our approach allows us to identify time points with greater activity of the predicted TFs, meaning time points where the number of exercise-regulated (differentially expressed) individual promoter regions is close to the cluster size (numerator and denominator opposite each point, respectively, in Fig. 4).
Fig. 4
TFs associated with co-expressed individual muscle promoter regions induced by exercise. Unsupervised analysis revealed 21 clusters of co-expressed individual promoter regions induced by exercise (b: before exercise). Each cluster includes data of 10 subjects and 4 time points; expression in each time point for each subject expressed by rank (the highest expression level: 4; the lowest: 1) and is presented as median and interquartile range. Denominator: the number of exercise-regulated individual muscle promoter regions at a time point; Numerator: the cluster size. The top enriched TFs are shown for each cluster
The findings are in good agreement with the data in the literature on exercise-induced activation of TFs regulating angiogenesis, mitochondrial biogenesis, and carbohydrate and fat metabolism in skeletal muscle [26]. Namely, nuclear receptors subfamily 4A (cluster 3), as well as estrogen receptors/estrogen-related receptors, peroxisome proliferator-activated receptor gamma and other nuclear receptors (clusters 11 and 13) were found to be associated with increased expression of the individual promoter regions at 3 h and 6 h after exercise, respectively.
On the other hand, our approach allows us to search for TFs (and their potential target genes) with roles in the regulation of stress-induced gene expression in human skeletal muscle that have been studied little or not at all. Thus, early response genes increasing expression at 1 h after exercise (clusters 2–3) were associated with multipotent transcription factors SRF, CREB-, ATF-, FOS-, and JUN-related. An increase in expression at 1–3 h of recovery was associated with calcium-dependent nuclear factor of activated T cells transcription factors and heat shock factors (cluster 3), TFs belonging to the ATF/CREB/AP-1 superfamily (clusters 2, 3, 8), as well as PAX-, NFKB-, POU, PAR-, SIX-, and Krüppel-related factors (KLFs, SPs) (clusters 4–8), while expression at the later stages of recovery was associated with muscle-specific factors MEF2 (cluster 9); NKX3, MAZ, NFE2L2, and paired-related homeodomain factors (cluster 9–13). The zinc finger superfamilies were activated in various combinations during the entire investigated period (clusters 2–8, 10, 11, 13, 16, 19, 21).
留言 (0)