
記住我

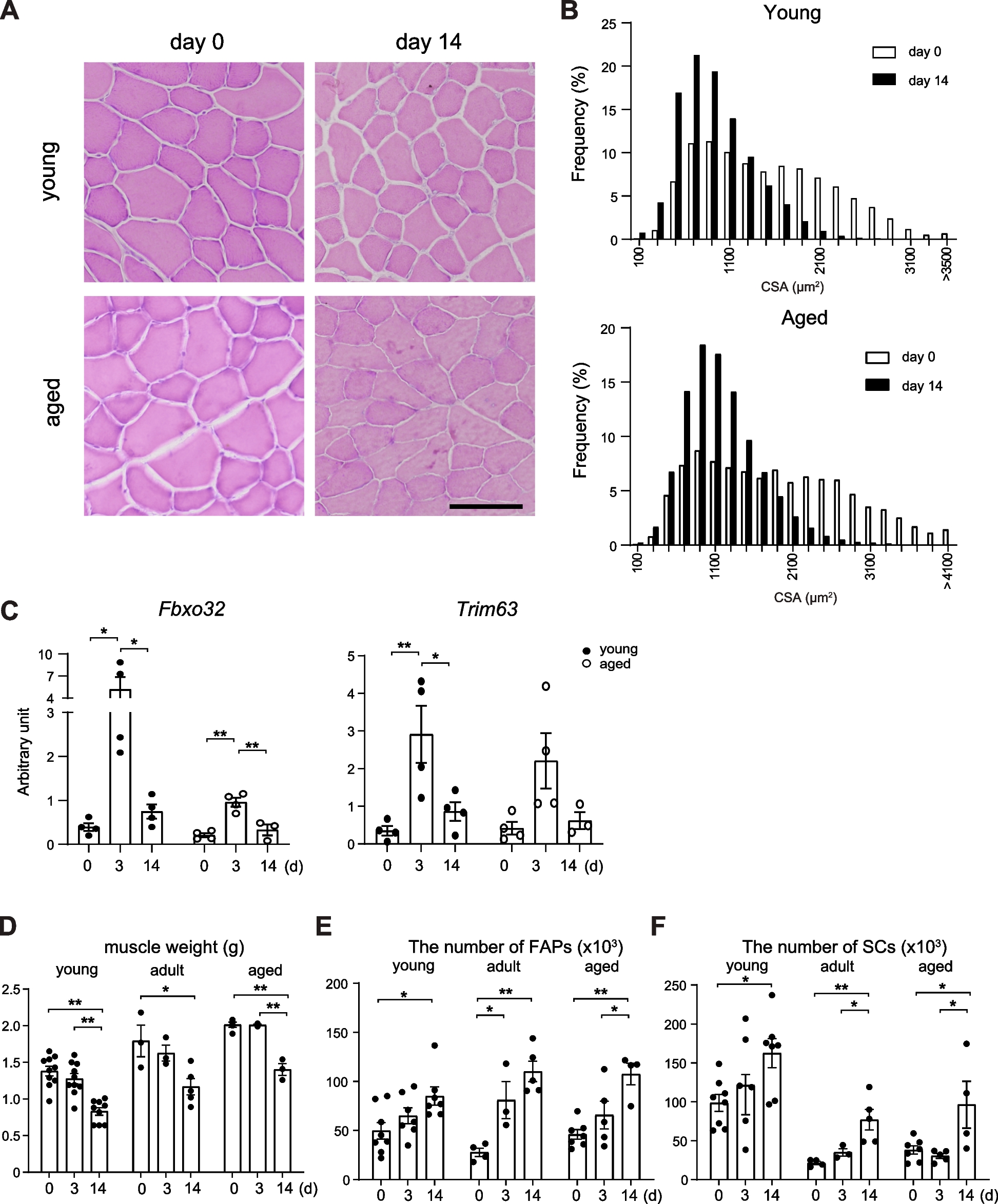




Skeletal muscle is a dynamic tissue that presents excellent regenerative ability and plasticity in response to external and internal changes, and both processes rely on myogenic-committed cells that reside in skeletal muscle, which are known as muscle satellite cells (MuSCs) [1,2,3,4]. MuSCs remain in a quiescent state under steady conditions [5] but start proliferating in response to damage or skeletal muscle loading. MuSC behavior is affected by multiple cell types, including myofibers, immune cells, and interstitial cells, including endothelial cells and mesenchymal progenitors (FAPs: fibro/adipogenic progenitors) (Figs. 1 and 2) [6,7,8,9,10,11]. Compared to the process of muscle regeneration, the mechanism regulating MuSC dynamics under muscle loading (such as a resistance training) has not been well investigated. However, recent studies have analyzed the mechanisms underlying MuSC proliferation and cell–cell communication in loaded muscles [9, 10, 12, 13]. We briefly summarize the process of muscle regeneration and load-dependent muscle hypertrophy according to key factors underlying MuSC behaviors and discuss six differences in MuSC dynamics and cell–cell interactions between the regeneration and hypertrophy processes.
Fig. 1
Process of skeletal muscle regeneration. When myofibers are damaged or dead, their debris is removed by inflammatory macrophages (M1 Mø). Using spaces and factors derived from macrophages and mesenchymal progenitors (FAPs), muscle satellite cells (MuSCs) actively proliferate (early stage). In the middle stage of regeneration, anti-inflammatory macrophages (M2 Mø) support the regulation of myogenic differentiation and nascent myofibers (myotubes), which grow to mature myofibers (late stage)
Fig. 2
Process of mechanical-loaded muscle hypertrophy. In unloaded muscles, Yap/Taz is distributed in the cytoplasm of mesenchymal progenitors. Mechanical loading induces nuclear localization of Yap/Taz in mesenchymal progenitors, and MuSCs subsequently proliferate beneath the basal lamina by the mesenchymal progenitor-derived factor thrombospondin-1 (Thbs1). Proliferated MuSCs fuse with myofibers, which leads to an increased number of myonuclei. Notably, the new myonuclei are located in the peripheral position of myofibers
Muscle regenerationWhen a myofiber is damaged, MuSCs exit from the quiescent state and become activated and proliferate (Fig. 1). MuSC activation follows after the expression of myoblast determination protein 1 (MyoD) [14, 15]. Subsequently, genes that regulate the cell cycle are upregulated and the cells begin to proliferate. In vitro experiments have revealed that approximately 2 days are required for the first cell division of quiescent MuSCs [16]. The subsequent cell division rate, which is estimated to be 12 h in vitro, is considerably faster than the initial cell division rate [16]. In a cardiotoxin (CTX)-induced muscle injury model in C57BL/6 mice, day 3 after injury corresponds to the peak number of proliferating myoblasts (daughter cells of MuSCs) [17] while day 4 after injury corresponds to the abundant production of immature myofibers called myotubes, which are generated by myoblast–myoblast and myoblast–nascent myotube fusion (Fig. 1). Active myoblast-dependent myotube growth occurs from days 5–10 postinjury [17]. During this process, MuSCs present a self-renewal capacity that maintains the number of MuSCs and ensures their ability to repeatedly regenerate after future damages. Although the exact timing of MuSC self-renewal is unknown, studies have suggested that self-renewal determination occurs in the mid-regeneration period (approximately 4–5 days after injury) [17, 18]. In addition, Pawlikowski et al. reported that the majority of MuSC self-renewal occurs between days 5 and 7 postinjury since EdU-labeling experiments demonstrated that the last MuSC division occurred during regeneration in this period [19]. In mice with CTX-induced muscle injury, myofibers are rebuilt to their original size approximately 2–3 weeks after injury (Fig. 1).
For efficient MuSC proliferation and differentiation, inflammatory cells, mesenchymal progenitors, and basal lamina are required [7, 20,21,22]. The infiltration of neutrophils is observed within 12 h [23], although the primary infiltrating cells subsequently shift to macrophages, which clean up dead myofibers [24]. Consequently, the space for MuSC proliferation is ensured. In the early stages of muscle regeneration, inflammatory M1 macrophages are the main subset, and they are then replaced by anti-inflammatory M2 macrophages (Fig. 1) [25]. Both macrophages and mesenchymal progenitors contribute to MuSC proliferation [6, 7, 24]. Similar to MuSCs, the number of mesenchymal progenitors and macrophages reach their peak approximately 3 days after CTX injection and then return to their original number [26].
Muscle hypertrophyThe crucial feature of overload-induced skeletal muscle hypertrophy is an increase in myofiber size, which requires two events: an increase in protein synthesis and then an increase in myofiber nuclei [27, 28]. The insulin-like growth factor 1 (IGF1)-Akt-mammalian target of rapamycin (mTOR) pathway is a well-known protein synthesis pathway. Akt also suppresses the Forkhead box O (Foxo) transcription factor, thereby inhibiting the ubiquitin–proteasomal and autophagic/lysosomal pathways [29, 30]. Other pathways, such as calcium signaling, have also been reported to activate mTOR [31], and these pathways were extensively summarized in our recent review [28].
An increased number of myonuclei by MuSCs is also required for efficient muscle hypertrophy (Fig. 2). Mice depleted of MuSCs did not exhibit increases in myonuclei [3, 32], indicating that myonuclei accretion, as well as myofiber generation, absolutely depends on MuSCs [1, 2]. The need for an increased number of myonuclei in muscle hypertrophy or MuSCs had been debated for two decades [3, 33, 34]. Although experimental conditions or methodologies may obscure the effect of increased myonuclei on the efficiency of muscle hypertrophy over relatively short-term (2–3 weeks) periods after surgical mechanical loading [9, 32], all recent studies have demonstrated that an increase in the myonuclei number is critical for long-term (>8 weeks) muscle hypertrophy [9, 28, 35]. Moreover, the increased myonuclear number and protein synthesis during muscle hypertrophy are coordinated because the disruption of new myonuclear accretion in overloaded muscle results in reduced Akt activation and downstream signaling [36]. Collectively, the data indicate that myonuclear accretion is required for sustained functional growth.
In our tenotomy-induced overloaded model, MuSCs started to express Ki67 at 2 days and substantial MuSC proliferation was observed approximately 4 days after surgery. Although the number was small, new MuSC-derived myonuclei were detected 4 days after tenotomy, and the number of MuSC-derived myonuclei gradually increased at least 2 weeks after surgery [12]. In loaded muscle, MuSC proliferation and differentiation seemed to occur concurrently (Fig. 2) [12, 37].
Compared with skeletal muscle regeneration processes, studies on cell–cell interactions during loading-dependent muscle hypertrophy are limited because the role of MuSCs is linked to their regenerative capacity, even in loaded muscle [37]. Undeniably, exercise is a widely accepted model of skeletal muscle loading that damages myofibers, particularly in rodent models [38]. Equivalent or similar experimental models have also been used to study the signaling pathways involved in the hypertrophy of living myofibers. Notably, the term “damaged myofiber” is based on physiological events (damage to the myofibril structures and damage to the myofiber sarcolemma with or without myofiber death; thus, in this study “damage” refers to “damage causing myofiber death”) [37]. In our surgical overload model, the areas of dead myofibers were rare, many living myofibers could be easily isolated, and MuSC proliferation on myofibers was observed from the loaded muscle, indicating that MuSC behaviors in the loaded muscles are regulated by different pathways compared with the regeneration process [12]. Collectively, these results led us to speculate on the differences in MuSC dynamics, cell–cell interactions, and their mechanism between muscle regeneration and hypertrophy processes (Figs. 1 and 2). Six differences are discussed below based on a comparison of the processes of muscle regeneration and hypertrophy among recent and other published studies.
Different activation and proliferation factorsMyofibers provide a specialized environment for MuSCs to sustain their undifferentiated and quiescent state. Notably, simply detaching MuSCs from myofibers may cause their activation and associated gene expression changes. Machado et al. demonstrated that the expression of early response genes, such as Jun, Egr1, and Fosb, is quickly upregulated in isolated MuSCs during cell preparation compared to that of bona fide quiescent MuSCs [39]. Therefore, during muscle regeneration, the loss of myofibers or factors secreted from damaged myofibers induce the activation and proliferation of MuSCs. For example, the secretion of tenascin-C [40] or GAPDH [41] from dead myofibers has been shown to induce the activation and proliferation of MuSCs. Several macrophage-derived factors (TWEAK, GFD3, GDF15, and IGF1) have been identified as regulators of MuSC proliferation and differentiation [42,43,44]. Mesenchymal progenitors also express factors that promote MuSC proliferation [6], including a matricellular protein named WISP1 (WNT1 inducible signaling pathway protein 1, also known as Ccn4), whose downregulation during aging is involved in the reduced proliferation of MuSCs in aged mice [45]. In addition, mesenchymal progenitors are critical for the infiltration of hematopoietic cells, including macrophages, into damaged muscles [6]. Collectively, the interplay among mesenchymal progenitors, macrophages, and MuSCs is critical for the successful progression of muscle regeneration.
On the other hand, the environment of MuSCs in overload-dependent muscle hypertrophy is not significantly altered because it does not accompany myofiber loss (Fig. 2) [37, 46]. Several mechanisms may be responsible for inducing the activation and proliferation of MuSCs:
(1)Edema observed in early overloaded muscles [47]
(2)Direct mechanical forces acting on MuSCs
(3)Factors leaked from myofiber wounds that are not involved in cell death
(4)Factors secreted from myofibers in a mechanical force-dependent manner
Additional factors may also be considered. Recently, in surgically overloaded plantaris muscles, we found that mesenchymal progenitors are critical for efficient muscle hypertrophy by regulating MuSC proliferation (Fig. 2) [9]. In this model, an initial, likely edema-induced, increase in muscle weight in mesenchymal progenitor-depleted mice was comparable to that observed in control mice. Meanwhile, the activation and proliferation of MuSCs were severely impaired by the loss of mesenchymal progenitors, suggesting that edema is unlikely to induce MuSC activation and proliferation [9]. In addition, the ability of MuSCs to directly sense mechanical forces should be similar in control and mesenchymal progenitor-deficient mice, making it unlikely that (1) and (2) alone would induce MuSC proliferation. Although strictly distinguishing (3) from (4) may be difficult, the ability of myofiber-derived factors to affect the proliferation and dynamics of MuSCs in loaded muscles has been clarified. Myofiber-derived IL-6 has been well investigated as an exercise-dependent factor that promotes MuSC proliferation [48, 49]. Succinate acid from exercised myofibers also affects MuSC gene signatures [50]. The relevance of mesenchymal progenitors and myofibers should be further investigated to reveal the entire mechanism that regulates MuSC activation and proliferation in loaded muscles.
We also found that mesenchymal progenitors secrete various growth factors in response to increased mechanical force via Yap1/Taz (Fig. 2), which are known as mechano-transducers [9]. In particular, we demonstrated that thrombospondin-1 (Thbs1), a member of the matricellular protein family derived from mesenchymal progenitors through Yap1/Taz, promotes MuSC proliferation by stimulating CD47 expressed on MuSCs in loaded muscle (Fig. 2) [
留言 (0)