
記住我



The vasculitides are a broad category of diseases characterized by inflammation of the blood vessels. Although the underlying etiology of these diseases is not always well understood, inflammatory changes and necrosis of the vessel walls can result in a variety of complications including aneurysm and pseudoaneurysm formation, vessel rupture with the potential for life-threatening hemorrhage, and compromise in the vascular lumen resulting in altered hemodynamics [1, 2]. CT and MR angiography have become important imaging tools due to their non-invasive nature and excellent visualization of the vessel wall and lumen [2]. Recognition of characteristic patterns of vascular involvement can help guide the diagnosis of the underlying vasculitis and ultimately lead to the initiation of appropriate treatment to help reduce the risk of significant complications.
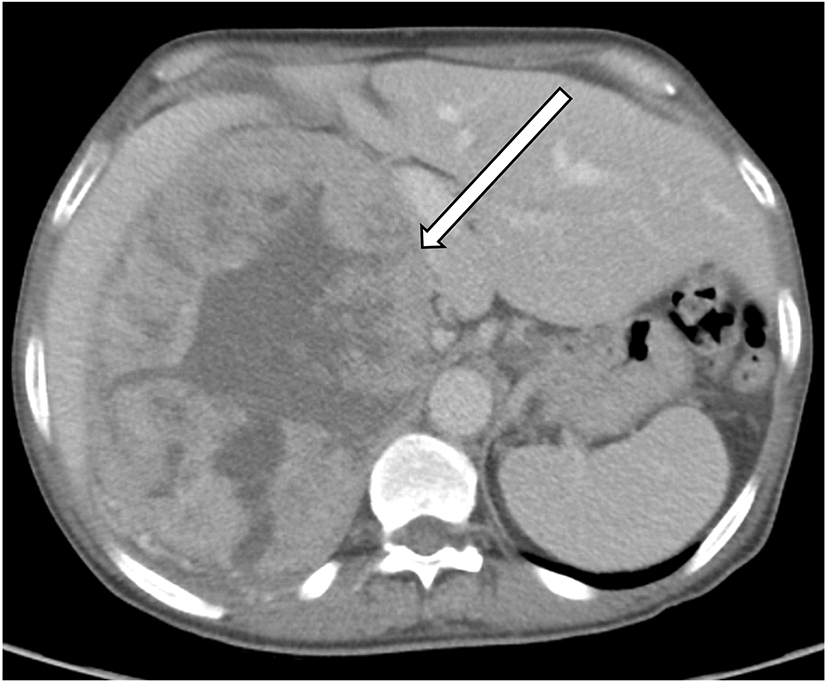
Systemic lupus erythematosus and drug-induced lupusSystemic lupus erythematosus (SLE) is a multi-systemic inflammatory process that results in damage to a variety of tissues secondary to antibodies and immune-complex mediated pathways, which can in turn lead to organ dysfunction involving different systems [3]. As with a range of inflammatory conditions, renal involvement is relatively common with SLE. The presence of vasculitis is well described in patients with SLE. While vasculitis in these patients often affects the gastrointestinal tract, it may also involve a variety of other organs [3]. Within the abdominal vasculature, lupus vasculitis may result in the formation of aneurysms which can lead to visceral arterial rupture [3]. These aneurysms may also affect the renal vasculature, and rupture of involved renal vessels leading to perinephric hematoma in the setting of lupus vasculitis is known as Wunderlich syndrome [4]. Lupus may be an idiopathic condition, although in several cases drug-induced lupus has been described. With certain medications, such as hydralazine, a continuum between drug-induced vasculitis and lupus has been proposed [5]. Therefore, recognition of lupus-related complications on imaging is important to help guide clinicians to the correct diagnosis so appropriate treatment can be initiated (Figs. 1 and 2).
Fig. 1
49-year-old male with multiple medical problems found to have laboratory evidence of drug-induced lupus. Axial (a) and coronal maximum-intensity projection (MIP) contrast-enhanced CT images (b) demonstrate innumerable aneurysms of varying sizes involving several renal artery branches bilaterally. This patient expired 9 days after this CT scan
Fig. 2
35-year-old male who presented with abdominal pain, found to have lupus nephritis on renal-biopsy. Axial wide (a) and narrow (b) field-of-view images of a contrast-enhanced CT demonstrate a filling defect within an expanded left renal vein (white arrow), consistent with acute renal vein thrombosis. The normal right renal vein is shown for comparison
Polyarteritis nodosaPolyarteritis nodosa (PAN) is a necrotizing vasculitis that involves small to medium vessels in a variety of organ systems [6]. Involvement of the renal vasculature has been reported in the majority of patients with PAN (ranging from 80–100%) [7]. Because of fibrinoid medial necrosis involving the media within the arcuate arteries, renal infarcts are often reported with PAN, as well as additional findings of aneurysm formation with or without rupture leading to retroperitoneal hemorrhage [8]. While multidetector CT angiography can often demonstrate findings of PAN, conventional angiography is superior at revealing microaneurysms that may be present in the disease process. PAN is typically responsive to therapy with corticosteroids and cyclophosphamide, with remission or cure reported in up to 90% of patients receiving these treatments [8] (Figs. 3 and 4).
Fig. 3
58-year-old female with a history of PAN and report of incidentally noted visceral aneurysms on outside imaging. Axial (a) and coronal MIP (b) contrast-enhanced CT images demonstrate aneurysmal dilation of a branch of the right renal artery
Fig. 4
Adult male patient with a history of PAN. Invasive angiographic images of the right (a) and left (b) kidneys demonstrate classic “micro-aneurysms” (white arrows) of the renal artery branches characteristic of PAN
Granulomatosis with polyangiitisGranulomatosis with polyangiitis (GPA, formerly known as Wegener’s granulomatosis) is a systemic necrotizing granulomatous vasculitis, often characterized by pulmonary symptoms in conjunction with renal dysfunction due to involvement of both systems [9]. Renal involvement is extremely common in the disease process, occurring in greater than 80% of reported cases of GPA [9]. Reported imaging features of GPA are variable, including renal scarring as well as solitary or even multiple renal masses [9, 10]. Like many vasculitides, GPA is highly responsive to steroids and cyclophosphamide [9] (Fig. 5).
Fig. 5
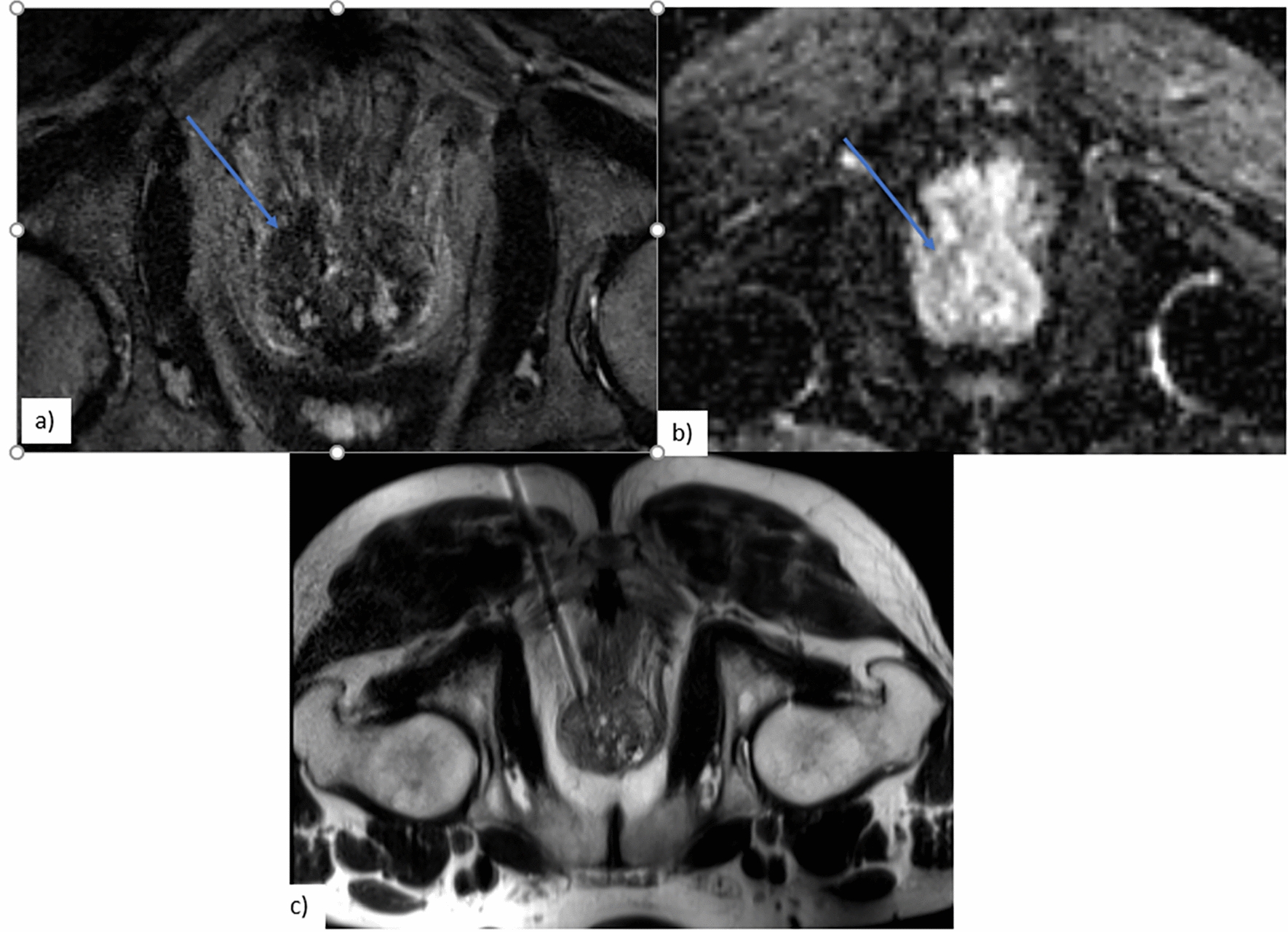
Coronal T2 (a) and T1-VIBE post-gadolinium (b) images demonstrate numerous small, hypointense bilateral renal cortical defects in a patient with renal-biopsy-proven GPA. Axial post-contrast CT( c) in another patient demonstrates a heterogeneous mass in the left kidney with central areas of low attenuation (white arrow) suggestive of necrosis. Biopsy confirmed the presence of non-caseating granulomas consistent with GPA
Leukocytoclastic vasculitisLeukocytoclastic vasculitis (LCV) is an inflammatory disorder involving post-capillary venules, generally thought to be the result of a hypersensitivity reaction [11, 12]. The disease can have manifestations in a wide variety of organ systems including the skin, mucous membranes, lungs, gastrointestinal tract and kidneys [11, 12]. Because cutaneous involvement is common, often presenting as palpable purpura, formal histopathologic diagnosis of LCV is often obtained via skin biopsy [12]. Renal involvement is common in LCV, with reported rates of necrotizing glomerulonephritis in up to 90% of cases [11]. Because the process involves small vessels, angiographic imaging may not directly demonstrate vascular complications such as aneurysms or pseudoaneurysms [11]. However, inflammatory changes within the involved organ system may be appreciated [11]. As with many vasculitides, LCV is commonly treated with steroids and immunosuppressant medications [12] (Fig. 6).
Fig. 6
50-year-old male with biopsy-proven cutaneous LCV. Coronal (a) and axial (b) post-contrast CT images demonstrate multifocal peripheral wedge-shaped areas of renal cortical hypoattenuation and parenchymal loss bilaterally (white arrows) consistent with scarring. This patient had normal-appearing kidneys on a PET-CT 7 months prior
Takayasu arteritisTakayasu arteritis is a chronic inflammatory vasculitis that involves large arteries, often the aorta and its major branch vessels [13]. The disease is most commonly seen in young women in the second and third decade of life, and is classified into six subtypes, depending upon the sites of involvement, with types III – V involving the abdominal aorta and its branches with varying degrees of involvement of the thoracic aorta [13]. Renovascular involvement in Takayasu arteritis is a commonly reported feature, with CT findings often showing concentric mural thickening as well as luminal irregularity and narrowing which can contribute to significant stenosis [13]. Collateral vessels may also be seen as a result of hemodynamically significant stenosis [13]. Recognition of typical imaging features of Takayasu arteritis is essential, as appropriate treatment (such as corticosteroid administration) can help to prevent potentially catastrophic complications of the disease process, such as aneurysmal rupture and critical end-organ ischemia. In addition to management with corticosteroids, interventional management (such as percutaneous transluminal balloon angioplasty) and surgical bypass may be indicated in certain cases where hemodynamically significant stenosis is present [13] (Figs. 7 and 8).
Fig. 7
42-year-old female with known Takayasu arteritis. Axial (a) and coronal (b) post-contrast MR angiogram MIP images demonstrate focal severe stenosis (white arrows) of the proximal right renal artery. Mild irregularity of the proximal left renal artery (white arrowhead) is also present. Sagittal post-contrast subtracted MIP (c) demonstrates diffuse irregularity of the descending thoracic and abdominal aorta (white arrows)
Fig. 8
Graphic depicting the six subtypes of Takayasu arteritis. The red shading indicates areas of involvement. Note that the paired vessels at the inferior aspect of the illustrated aorta represent the renal arteries
Immunoglobulin G4 vasculitisImmunoglobulin G4 (IgG4)-related disease is a multisystemic disease process related to infiltration of tissues by plasma cells and lymphocytes positive for IgG4, which can result in a variety of clinical manifestations [14]. The most well-known and classic association with IgG4-related disease is autoimmune pancreatitis, although additional sites of involvement (including renal involvement) are becoming more well recognized [14]. Renal involvement has been described in up to 35% of patients with autoimmune pancreatitis, and can have several different imaging manifestations, the most common being peripheral wedge-shaped cortical-based lesions, and less commonly a rim of tissue surrounding the kidney(s), diffuse patchy involvement, renal sinus nodules, and wall thickening of the renal pelvis [14]. Direct arterial involvement can also be seen with IgG4-related vasculitis with renal involvement. IgG4 disease is typically highly responsive to treatment with corticosteroids [14] (Figs. 9 and 10).
Fig. 9
33-year-old male patient with confirmed IgG4 vasculitis. Coronal post-contrast CT images (a) and (b) demonstrate ill-defined, hypoenhancing soft tissue surrounding the right renal artery (white arrows) consistent with inflammation. Coronal post-contrast CT nephrogram (c) demonstrates a large, wedge-shaped cortical defect in the right kidney consistent with infarct. Coronal CT angiogram (d) shows resolution of the inflammation following steroid therapy
Fig. 10
72-year-old male with acute kidney injury on a background of chronic kidney disease, with biopsy demonstrating IgG4 vasculitis. Axial T2 with fat saturation (a) and post-contrast GRE (b) demonstrate multifocal areas of renal cortical irregularity and scarring bilaterally
Behçet’s diseaseBehçet’s disease is a multisystem inflammatory vasculitis. While classically reported symptoms include oral and genital ulcers and uveitis, involvement is not confined to these areas, and multiple organ systems can be involved, including the venous and arterial systems, with the venous system being more commonly involved (accounting for approximately 85% of vascular involvement) [15]. Venous complications most commonly include thrombophlebitis, and less frequently venous occlusion and aneurysm formation [15]. Arterial vascular manifestations of Behçet’s disease include arterial aneurysms and pseudoaneurysms, thought to be the result of vasa vasorum inflammation resulting in elastic fiber destruction leading to vascular dilation [15, 16]. Additional arterial manifestations include stenosis and occlusion [16]. When encountered in the renal arterial system, areas of stenosis and occlusion can result in renal infarction. As with those of many of the vasculitides, manifestations of Behçet’s disease are often responsive to steroid therapy [16] (Fig. 11).
Fig. 11
Coronal post-contrast MRI (a) in a patient with Behçet’s disease demonstrates narrowing of a branch of the main renal artery (arrow) with associated cortical defect consistent with infarct. Axial contrast-enhanced CT (b) demonstrates an area of diminished right renal perfusion (arrow) consistent with infarct. Axial contrast-enhanced CT 1 month later (c) demonstrates evolving infarct (arrow) in the right kidney
留言 (0)