
記住我


All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee at which the studies were conducted and with the Helsinki Declaration and comparable ethical standards, Good Clinical Practice guidelines, and other regulatory requirements. The study was approved by the institutional review board of each participating center. All subjects provided written informed consent before participating in the study.
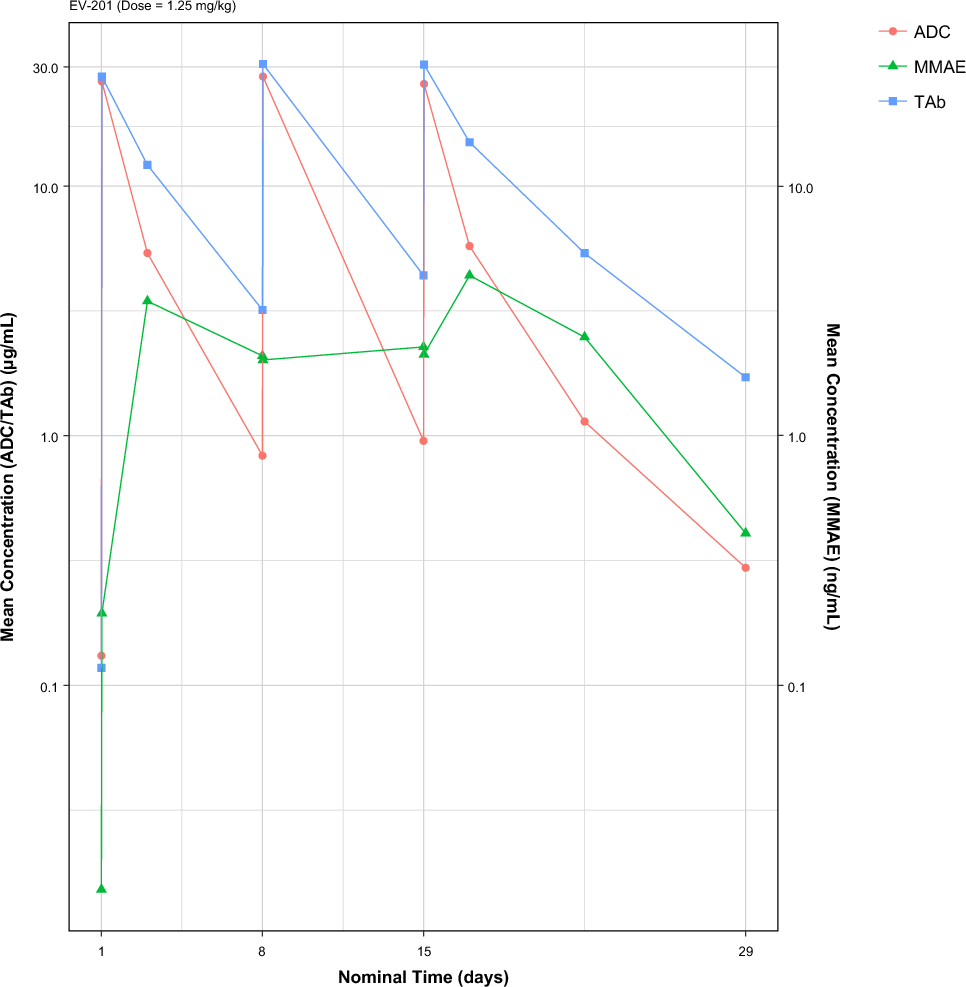
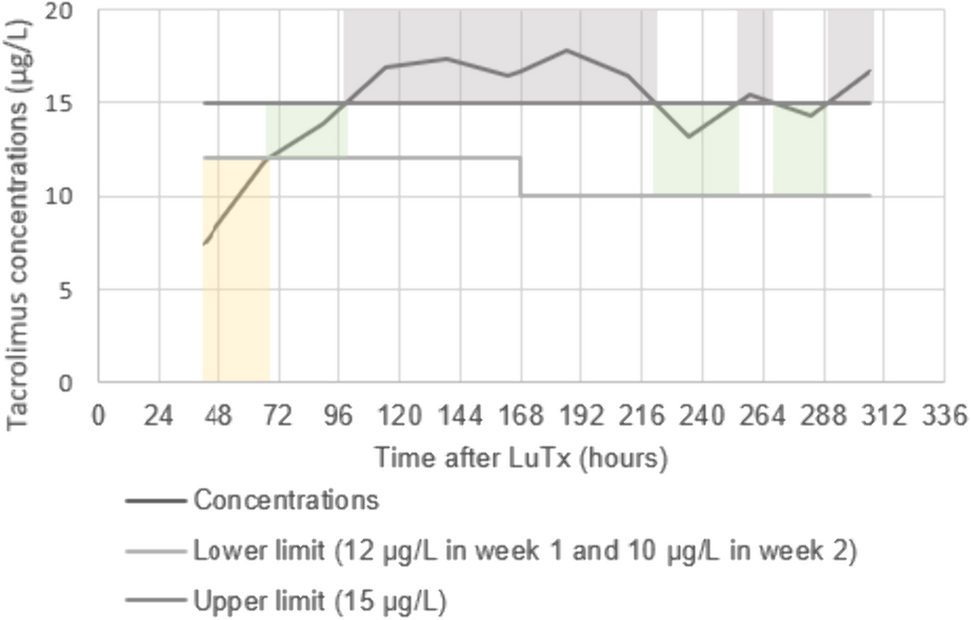
Study DesignThis study was conducted as a phase I/II, multicenter, randomized, placebo-controlled, double-blinded, dose-escalation study. At the screening test, patients who met all inclusion criteria and did not meet any exclusion criteria were randomly assigned via a centralized randomization system to receive either upacicalcet or a placebo. This study consisted of a single-dose study and a 3-week multiple-dose study. The dose range of upacicalcet in this study was 0.025–0.8 mg, which was considered to be effective, safe, and well tolerated based on the results of the phase I study. The study design is illustrated in Fig. 1.
Fig. 1
Study design and the target number of patients
For the single-dose study, six patients in each step cohort were enrolled in seven-dose steps and then randomized 2:1 to receive upacicalcet or a placebo. The first-dose step cohort (Step 1) received a 0.025 mg intravenous injection of upacicalcet. After the safety of the Step 1 cohort was confirmed, the second cohort study was initiated. Each subsequent cohort was subjected to the same procedure. Criteria for consideration to each step-up are shown in the ESM.
For the 3-week multiple-dose study, 12 patients in each step cohort were enrolled in three-dose steps and randomized 3:1 to receive upacicalcet or a placebo. The upacicalcet dose for the first multiple-dose step cohort (Step 1) was 0.05 mg. After completing the safety confirmation with the single dose of 0.1 mg, the first multiple-dose step cohort study was initiated. Subsequent multiple-dose step cohort studies were initiated after safety confirmation of both the previous multiple-dose cohort and a higher single-dose cohort than the planned multiple-dose study. Patients who participated in the one step were not allowed to participate in another cohort study.
Inclusion and Exclusion Criteria and RandomizationParticipants who met all the inclusion criteria and did not meet any exclusion criteria at the screening test were enrolled in the study. The inclusion criteria were as follows: age between 20 and 80 years; hemodialysis or hemodiafiltration three times a week for at least 12 weeks prior to the screening test; serum intact PTH (iPTH) level > 240 pg/mL; serum corrected Ca2+ (cCa2+) level ≥ 8.4 mg/dL; and ability to provide written informed consent. The main exclusion criteria were as follows: primary hyperparathyroidism, received cinacalcet or changed the dosage and administration of active vitamin D preparation, Ca preparation, or P adsorbent from 2 weeks prior to the screening test. The other inclusion and exclusion criteria are shown in the ESM. The participants were randomly assigned according to an assignment form prepared by the external organization for case registration (EPS Corporation, Tokyo, Japan).
InterventionsIn the single-dose study, allocated subjects were admitted to the hospital the day before or on the day of dosing, subject eligibility was finally confirmed, and upacicalcet or placebo was administered intravenously on day 1 as slowly as possible between 2 and 4 h after the end of dialysis on the day of study drug administration. Patients were hospitalized until day 4. A post-treatment examination was performed on day 8. The planned doses of upacicalcet or placebo were 0.025, 0.05, 0.1, 0.2, 0.4, 0.6, and 0.8 mg.
In the multiple-dose study, allocated subjects were administered with upacicalcet or placebo on the first dialysis day of the week (after the maximum dialysis interval). The study drug was administered intravenously into the venous side of the dialysis circuit immediately before the end of hemodialysis. The dosing interval and duration were three times per week for 3 weeks until day 22. A post-treatment examination was performed on day 29. The planned doses of upacicalcet or placebo were 0.05, 0.1, and 0.2 mg.
Pharmacokinetic EvaluationsTo determine the plasma concentrations of upacicalcet and its predominant metabolites (M1, M2, and M3), blood samples were collected at the following timepoints: in the single-dose study, before administration, and 10 and 30 min, and 1, 3, 6, 18, 30, 42, 66, 67, and 70 h after administration; in the multiple-dose study, day 1, 3, 5, 8, 10, 12, 15, 17, 19, and 22 (before dialysis); day 1 (10 and 30 min, and 1 h after administration); day 17 (10 and 30 min and 1 and 20 h after administration); and day 19 (before administration).
Blood samples were collected in a vacuum blood collection tube containing 2 mL of sodium heparin (Insepack II-D, Sekisui Medical Co., Ltd. Tokyo, Japan) at each timepoint. The collected blood was immediately centrifuged, and the separated plasma samples were frozen for storage and transported to Shin Nippon Biomedical Laboratories, Ltd. (Tokyo, Japan). The concentrations of upacicalcet and its metabolites in the plasma were measured by liquid chromatography-tandem mass spectrometry. The pharmacokinetic parameters in the single-dose study were calculated as follows: maximum (peak) plasma drug concentration (Cmax), time to Cmax, area under the plasma concentration–time curve from zero to 66 h, area under the plasma concentration–time curve from time zero to infinity, elimination half-life, mean residence time from zero to time t, mean residence time to infinity, apparent total body clearance of drug from plasma, apparent volume of distribution at steady state, removal rate by hemodialysis, and hemodialysis clearance. The formulas for removal rate and clearance by dialysis are shown in Table 3. In the multiple-dose study, the Cmax and removal rate by hemodialysis were calculated.
Pharmacodynamic EvaluationsAs upacicalcet is a calcimimetic developed for the treatment of SHPT, the pharmacodynamic parameters of upacicalcet determined were serum iPTH, serum cCa2+, and P levels. The Payne’s formula was used to calculate cCa2+. As exploratory measurements, we measured whole PTH, fibroblast growth factor 23, 1,25(OH)2 vitamin D, ionized Ca2+, and calcitonin levels in the single-dose study and tartrate-resistant acid phosphatase-5b, bone-specific alkaline phosphatase, and osteocalcin, which are markers of bone metabolism, in the multiple-dose study.
Blood samples were collected in vacuum blood collection tubes (VP series, Terumo Corporation, Tokyo, Japan). Blood samples for the pharmacodynamic parameters were collected at the following timepoints: in the single-dose study, before dialysis, before administration, and 10 and 30 min and 1, 3, 6, 18, 30, 42, 66, and 70 h after administration; in the multiple-dose study, day 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, and 29 (before dialysis); day 1 (30 min and 1 h after administration); day 17 (30 min and 1 and 20 h after administration); and day 19 (before administration) and were measured by SRL Medisearch Inc. (Tokyo, Japan).
Safety EvaluationsFor all adverse events (AEs) that occurred after the start of the study treatment, the occurrence and incidence of AEs and adverse drug reactions (ADRs) were summarized for each treatment period and dose. The incidence of AEs and ADRs for each treatment period and dose was summarized using the system organ class and preferred term using Medical Dictionary for Regulatory Activities J version 18.1.
The safety and tolerability profiles of upacicalcet were assessed in terms of occurrence of AEs, vital signs, clinical laboratory measurements, and ophthalmology examination (multiple dose only). Vital signs were monitored at each institution, and 12-lead electrocardiograms were monitored and analyzed by Suzuken Co. Ltd. (Nagoya, Japan).
Statistical AnalysisPatients who received placebo were evaluated collectively as the placebo group without distinguishing the step cohort. Patients who received upacicalcet were evaluated in each dosage group. Patients who experienced an accidental overdose were excluded from pharmacokinetic and pharmacodynamic analyses. The relationship between the measured QT interval as corrected by the Fridericia method (QTcF) and the change in QTcF before dosing and the concentration of upacicalcet and its metabolites (M1, M2, and M3) in plasma were examined for each dose group of upacicalcet by a regression analysis. The relationship between cCa2+ levels and QTcF was also examined in the same manner. SAS version 9.3 (SAS Institute, Cary, NC, USA) was used to perform other statistical analyses and to summarize the data.
留言 (0)