
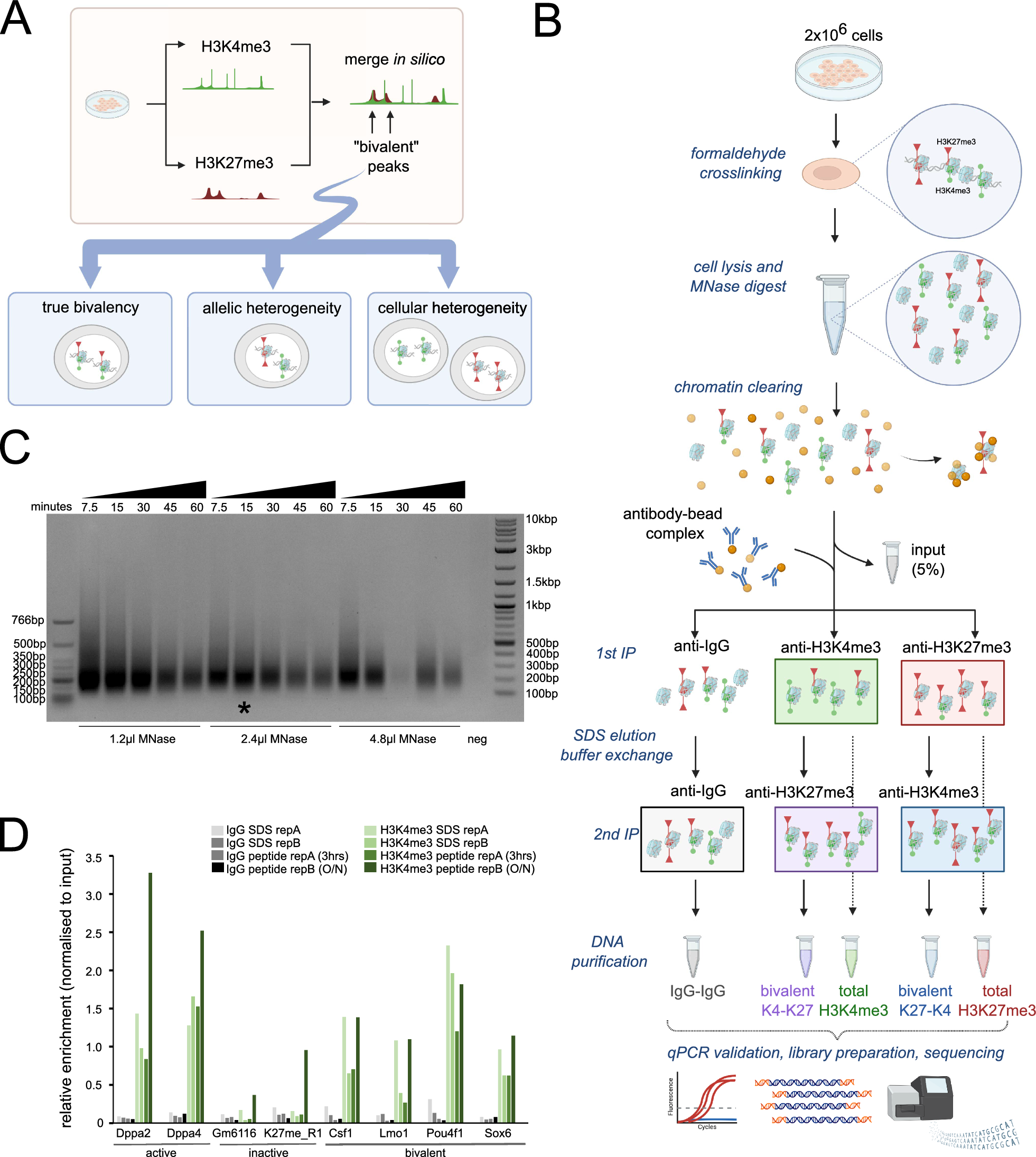
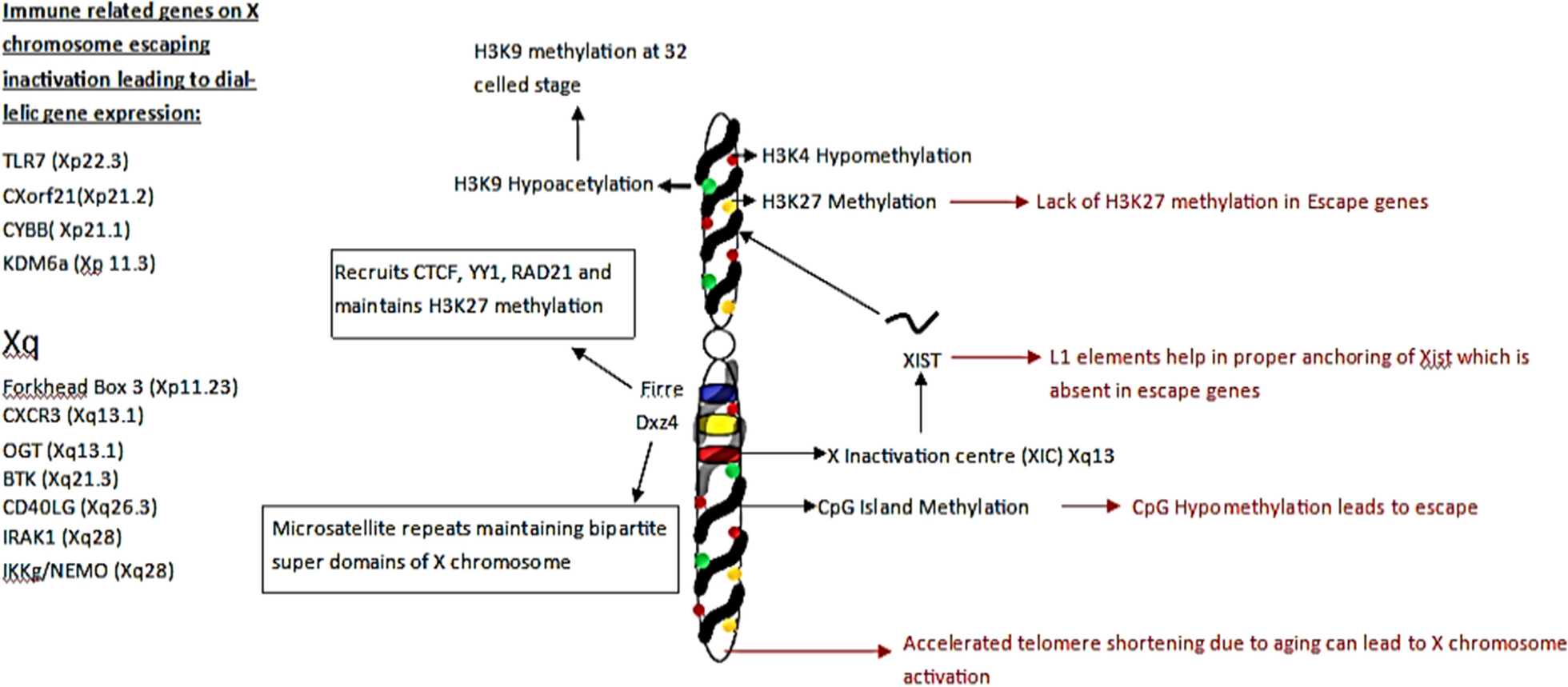
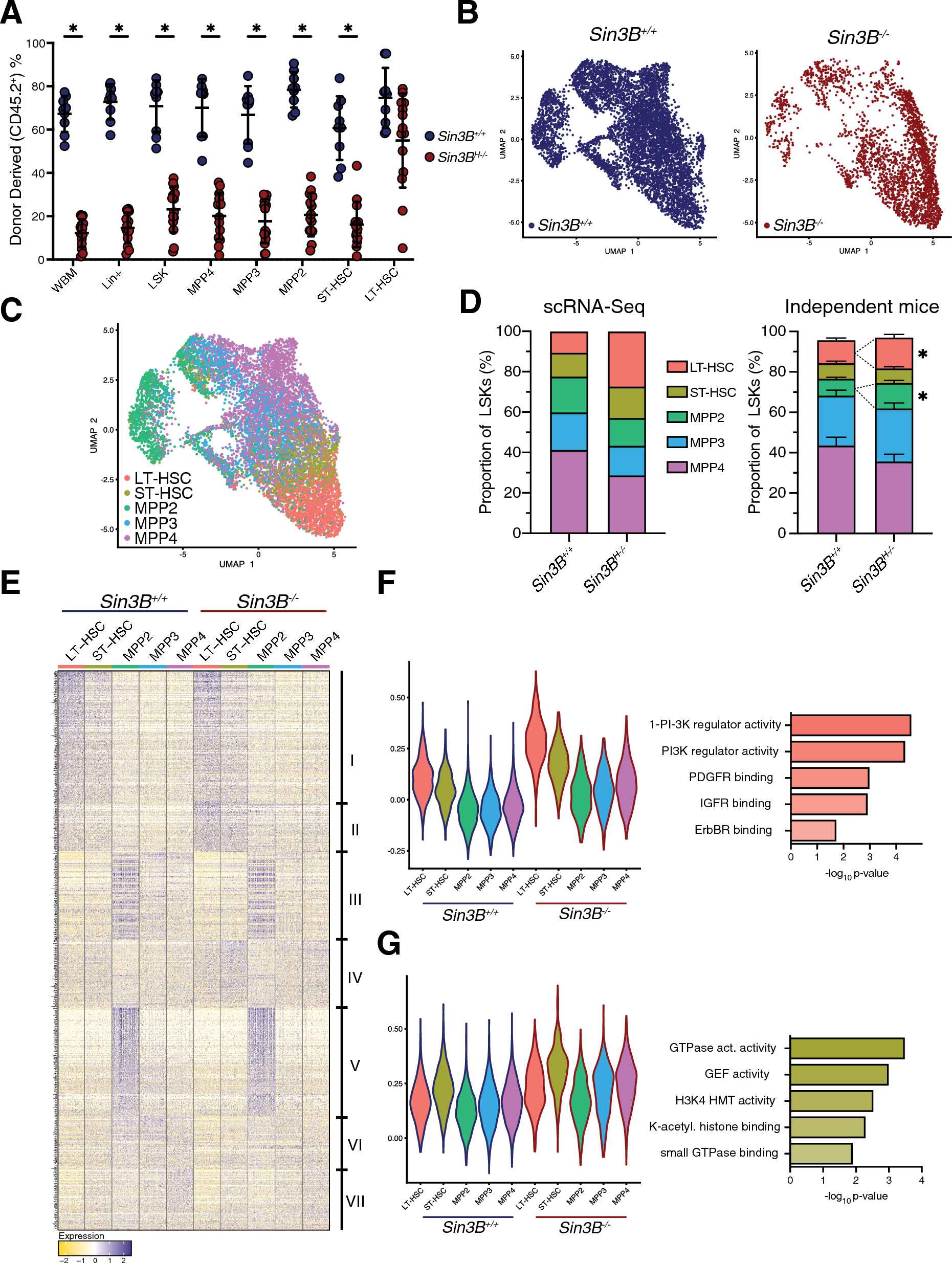
記住我



The mammalian genome is partitioned into A and B compartments, which correspond to transcriptionally active and silent chromatin, respectively [24]. Our previous studies showed that HMGN proteins bind to cell-type-specific regulatory sites, e.g., enhancers, super-enhancers, and assist in the regulation of gene transcription and cell identity [19, 20, 25]. However, whether HMGN protein enrichment in chromatin correlates with three-dimensional (3D) higher order chromatin structures is still unknown. In this study, we generated high-resolution in situ Hi–C data from three different cell types: mouse embryonic fibroblasts (MEFs), resting B (rB) cells, and induced pluripotent stem cells (iPSCs) [19]. We used Hi–C data of ESC cells from another study [26] and HMGN protein ChIP-seq data from our previous studies [19, 20]. We first identified and analyzed the A/B compartments in each of these cell types, using CScoretools [27]. Based on the A/B compartment identification results from CScoretools, the ratio of the genomic distance of compartment A to B is 4:5 in MEFs, 9.1:10 in resting B cells, 3:5 in ES cells, and 4:5 in iPSCs, suggesting that the total length of A and B compartments of the entire genome is similar in all of the cell types. Next, we integrated HMGN1/2 ChIP-seq data with Hi–C compartment analysis results. Our results show that HMGN proteins are located mainly within A compartments: in MEFs, about 75% of HMGN1 and HMGN2 peaks are enriched within the A compartment, and about 95%, 80%, and 80% HMGN peaks are in the A compartment in rBs, ES cells, and iPSCs, respectively (Fig. 1A). There is a sharp increase in HMGN signals across the boundaries between B and A compartments in all three cell types, as shown in Fig. 1B–E (left panels), which plot the average HMGN signals at a 200 kb window across all of the B → A boundaries. Our previous studies have shown that HMGN1 and HMGN2 signals are very similar across all the cell types. Therefore the results here are the average of HMGN1 and HMGN2. The B → A boundary regions of all four cell types exhibit stable and high mappability (75 bp read length and 2 mismatches allowed) of about 0.9 (Additional file 2: Fig. S2A), which excludes the possibility that the observed trend is influenced by any type of mapping bias due to sequence repeats. IGV genome browser snapshots (Fig. 1B–E, right panels) show individual examples of HMGN ChIP-seq signals highly enriched in the A compartment and that overlap with the active enhancer marker, H3K27ac. These results are consistent with our earlier studies that find that HMGN proteins are involved in active gene regulation [20, 28].
Fig. 1
HMGN proteins (HMGN1 and HMGN2) are enriched at A compartments. A Percentage of HMGN1 and HMGN2 ChIP-seq peaks located in A compartment in MEFs, rBs, ESCs, and iPSCs. B–E Left panels: average HMGN1 signals in 200 kb windows across boundaries from B compartment to A compartment in MEF, rB, ESC and iPSC cells. B–E Right panels: individual examples of HMGN ChIP-seq signals enriched in A compartment and overlap with active enhancer maker H3K27ac. Snapshots are made from IGV genome browser
Unaltered 3D chromatin structures upon HMGN protein depletionThe eukaryotic genome is organized into distinct functional domains with different scales and compaction levels [2]. To understand the effect of HMGN proteins on higher order chromatin structures, we performed Hi–C experiments on wild-type (WT) and HMGN1/2 double knockout (DKO; Hmgn1−/−; Hmgn2−/−) MEF, rBs, and iPSCs. The WT and Hmgn DKO mice, and cells derived from these mice have been extensively characterized. DNA sequence analyses show the absence of the deleted genomic sequences, RNA sequence analyses show the absence of transcripts of the deleted exons, and Western analyses show that DKO cells do not express HMGN1 and HMGN2 proteins [19, 20, 25, 47]. Next, we generated Hi–C contact matrix maps, using the HiC-Pro pipeline [29], and visualized these data in Juicebox [29, 30]. We evaluated the reproducibility of replicates with the method of HiCRep [31]. The stratum adjusted correlation coefficient (SCC), an indicator of similarity levels between Hi–C interaction matrices, showed that the SCCs between any two of the WT replicates range from 0.985 to 0.995, and the SCCs between DKO replicates are at a similar level. Interestingly, the SCCs as compared with any WT and DKO samples also are at the same range, from 0.985 to 0.995. SCCs between samples from different cell types range from 0.5 to 0.62 (Additional file 2: Fig S1). These data suggest that the depletion of HMGN proteins does not significantly alter 3D chromatin contact matrixes. Next, we quantified A/B compartment differences between WT and DKO cells with CScoretools [27]. The output of CScoretools is a compartment score (C-score) that reflects the chance of a given genomic window being in the A or B compartment. The genome is divided into bins of 25 kb (bin sizes of 10 kb, 50 kb, and 100 kb generate similar results). A C-score ranging from − 1.0 to 1.0 is calculated for each bin. A positive value means A compartment while a negative value means B compartment (illustrated in Additional file 2: Fig S2A). The C-scores of WT cells is plotted against DKO cells for all bins (Fig. 2A). The CScore results showed that HMGN protein depletion has little effect on compartment scores, genome-wide, in all three cell types (Fig. 2A). Although there are some sites that switch from B compartment in WT to A compartment in DKO MEF cells (dots in the left top square), further examination of these dots revealed that they are from mainly two regions, at Chr4 and Chr13 (Additional file 2: Fig. S2C), and we did not find any correlation between these changes and gene expression. We noticed that the A/B compartment score differences between WT and DKO cells are greater in iPSCs than in MEFs and rBs, which is probably attributable to the fact that induced pluripotent stem cells have more open chromatin and are more sensitive to cellular nucleus status changes [32, 33]. Figure 2B further exemplifies the similarity of contact matrixes between WT and DKO cells at 50 kb, 25 kb, and 10 kb resolution.
Fig. 2
Unaltered 3D chromatin structure between WT and HMGN1/2 DKO cells. A Comparison of CScore of WT and DKO cells in MEF, rBs, and iPSCs. CScores are calculated at 25 kb resolution. B Examples of Juicebox illustration of similar Hi–C chromatin structure between WT and DKO at specific regions, at resolutions of 50 kb, 25 kb, and 10 kb in MEF, rB, and iPSCs. C Examples of similar TAD boundaries between WT and DKO in MEF, rB, and iPSCs. Hierarchical TADs are marked with lines of different colors
TADs are considered the structural and functional units of mammalian genomes. TADs are characterized by a high frequency of intra-domain chromatin interactions but infrequent inter-domain chromatin interactions [34]. To investigate whether HMGN proteins affect TAD structures, we identified TADs in WT and DKO cells, using OnTAD [35]. Our results suggested that TAD structures remain intact in HMGN-depleted cells (Fig. 2C). Genome-wide, WT and DKO cells have a similar number of TADs with a comparable TAD size distribution (Additional file 2: Fig S3A, B). In summary, our results suggested that Hi–C contact matrices, A/B compartments, and TAD structures were largely unchanged upon HMGN protein depletion. Considering the highly conserved nature of TADs and higher order chromatin structures among different tissues or even species [36], it is not surprising that HMGN protein depletion causes no significant structural changes in higher order chromatin structures, as HMGs are not found in lower eukaryotes.
HMGN proteins occupy promoter interaction regions that are highly enriched for cis-regulatory featuresEnhancer–promoter interactions play a critical role in gene regulation. In particular, long-range cis-regulatory elements modulate target promoters through DNA looping and folding, a mechanism that bridges the distal enhancers to proximity to the target promoters [37]. Because our previous studies found that HMGN proteins bind to cell-type-specific regulatory sites [19], a related question is whether and how HMGN affects the spatial enhancer–promoter interactions in 3D nuclear space. To investigate this, we performed Promoter Capture Hi–C (PCHC) in WT and DKO MEFs, rBs, and induced pluripotent stem cells (iPSCs). The PCHC technique was developed to enrich promoter-containing ligation products from Hi–C libraries and to reduce the complexity of Hi–C libraries [38]. After mapping the readings with the HiCUP program [39], we identified significant promoter interaction regions (PIRs) by using the CHiCAGO pipeline with a threshold CHiCAGO score of ≥ 5 [40]. The numbers of significant PIRs from each experiment range from about 82,000 to 145,000, depending on the library sizes.
The enrichment analysis of the CHiCAGO program showed that the identified PIRs are highly enriched in HMGN protein ChIP-seq signals and other genomic features involved in active transcription regulation, including histone markers H3K4me3, H3K27ac, and H3K4me1, and protein ChIP-seq signals for p300 and CTCF. The fold enrichment for these features ranges from 1.9 to threefold, For the repressive histone markers, H3K27me3 is only slightly enriched in the identified PIRs, with fold enrichment of 1.15 in MEF and 1.25 in rB cells. It might be related to the role of Polycomb proteins in nuclear architecture [41]. H3K9me3 is depleted to 71% in iPSCs (Fig. 3A). For example, Fig. 3B shows that the promoter of the MEF specific gene Erc2 specifically contacts multiple genomic regions hundreds of kilobases away, both upstream and downstream, decorated with HMGN proteins and H3K27ac signals (Fig. 3B). There are, however, no significant similar contacts on those regions in rBs or iPSCs. Examples of cell-type-specific promoter–enhancer interactions and HMGN protein occupancy at PIRs in rBs and iPSCs are shown in Additional file 2: Fig. S4. Overall, our PCHC and ChIP-seq analysis reveal that HMGN proteins occupy cell-type-specific PIRs in the 3D genome.
Fig. 3
HMGN proteins bind to promoter interaction Regions (PIRs), which are highly enriched for cis-regulatory features involved in active transcription. A Chromatin features of promoter-interacting fragments detected with CHiCAGO. Yellow bars indicate overlaps of the genomic features with cis-interacting fragments within 1 Mb of promoter baits; blue bars indicate expected overlap values based on 100 random subsets of HindIII fragments. These subsets were selected to have a similar distribution of distances from gene promoters as the interacting fragments. Error bars represent 95% confidence intervals. Genomic features include ChIP-seq peaks of HMGN1/2, histone markers, CTCF, and p300. The difference between detected interactions and the expected value is significant for all genomic features with p value < 1e−30. The features involved in positive transcription regulation have a fold enrichment of ~ 2–3 while the repressive markers, H3K27me3 and H3K9me3, are either only slightly enriched or depleted. B Upper panel: snapshots of interactions with the Erc2 promoter identified with CHiCAGO in MEF cells. Lower panel: ChIP-seq signals of HMGN1, HMGN2 and H3K27ac in the same region. HMGN proteins specifically occupy at MEFs-specific site (Erc2) and its enhancers in MEF cells
We examined the statistically significant differential interactions in PCHC data between WT and DKO cells with Chicdiff [42]. We identified 131 differential interactions between MEF WT and DKO cells (Additional file 2: Fig. S5). These 131 sites, however, come from interacting regions with six gene promoters, for which there is no difference in gene expression levels between WT and DKO. We found no differential interactions in resting B cells or iPSCs. We thus determined that HMGN protein depletion has minor effects on promoter–enhancer interactions in the three different cell types, which is consistent with the Hi–C data analysis results that show that HMGN proteins do not directly regulate higher order chromatin structures.
Proteomic profiling identifies proteins associated with HMGN proteins in chromatinPrevious examination of HMGN binding sites in various cells demonstrated the presence of an active chromatin signature at many of its binding sites. This signature is defined as a co-localization with high H3K27ac, H3K9ac, H3K4me1, and H3K4me3 histone markers, various nuclear factors, and increased chromatin accessibility [19, 20]. Preferential HMGN association with the A compartment in this work confirms the postulation that HMGNs contribute to various nuclear activities involved in transcription regulation. Next, we addressed the question of whether HMGN proteins have a preference to be juxtaposed to specific nuclear factors.
We performed chromatin immunoprecipitation combined with mass spectrometry (ChIP–MS) using HMGN antibodies in two cell types (ESCs and MEFs) derived from WT and DKO mice. We aimed to identify protein factors associated directly with or neighboring HMGN1 and HMGN2 proteins on nucleosomes [42, 43]. The protocol consists of the following steps: crosslinking, sonication, and immunoprecipitation with HMGN1 and HMGN2 antibodies, protease digestion, and LC–tandem MS (LC–MS/MS) analysis. This is followed by a computational murine database peptide search and protein identification (Fig. 4A). We reversed the cross-linked sonicated chromatin and checked DNA size distribution using TapeStation (Fig. 4B, C). Our results show that the fragmented DNA is normally distributed, with a peak of 180 bp, which ensures that protein factors identified by the ChIP–MS procedure are directly associated with or neighbors of HMGN proteins on the same nucleosome.
Fig. 4
Chromatin profiling by ChIP–MS identifies proteins that reside nucleosome-long proximity to HMGN-occupied regions of chromatin. A Schematic overview of ChIP–MS assay. Mouse embryonic stem cells and MEF cells were cross-linked, and the chromatin was isolated, sonicated, and immunoprecipitated. DKO cells served as a negative control. Overall, there were 16 samples: two immunoprecipitations (HMGN1 and HMGN2), two cell types (ES and MEF), two genotypes (WT and DKO), and two biological replicates. Proteins, co-purified with HMGN and identified by UHPLC/MS/MS, represent HMGN binding partners. B ChIP DNA visualized by the Agilent TapeStation System after the optimized sonication step. More than 70% of total DNA fragments ranged from shorter than 505 bp to longer than 58 bp. C The table with the total number of identified proteins in individual samples. The number of proteins selected as HMGN binding partners with various (WT/DKO) cutoff limits in various cells
A successful ChIP–MS experiment generally results in the identification of 300–900 proteins, with 5–10% of these as specific binding partners [44]. All tests are abbreviated below as ES_N1, ES_N2, MEF_N1, and MEF_N2, and each test consisted of 4 samples: two WT and two DKO replicates. We observed 200–2000 protein factors per biological replicate (for a full list, see Additional file 1). All proteins identified by ChIP–MS analysis in DKO samples serve as false positive hits. Western analyses with antibodies to SMARCA5 and ATRX further verified that the ChIP MS data identified proteins that neighbor HMGN proteins. These proteins are detected in immunoprecipitates of WT cells but not in immunoprecipitates of the DKO control cells (Additional file 2: Fig. S6).
Two replicated average abundance values were calculated and served to establish a threshold measurement to identify true HMGN binding partners. The [WT/DKO] abundance ratio measures specific immunoprecipitation versus non-specific interactions. We defined proteins as specific HMGN binding partners if they have been preferentially identified in the target (WT) cells, either in negligible amounts or absent in the DKO cells. We considered that the criteria for a positive versus negative outcome would be the presence of protein in immunoprecipitated fractions of both WT biological replicates and that their average ((WT1 + WT2)/2) abundance was at least ten times higher than in DKO samples. We then identified 1157 factors with at least a tenfold difference in the abundance ratio, a measure for 1the amount of protein specifically immunoprecipitated, 833 factors at a 50-fold threshold, and 798 factors at a > 500-fold threshold (Fig. 4C). All subsequent analyses were conducted for proteins with WT/DKO ratios of > 10.
HMGNs juxtapose proteins that execute various DNA- and chromatin-based activitiesThe lists of proteins—HMGN binding partners—in each of four groups of experiments are presented in Additional file 3: Table S1 (tabs indicate experiment groups). The proteins are shown with their average abundance value in HMGN-immunoprecipitated material. The abundance fluctuations may underlie distinct functional characteristics of the different cell types and various HMGNs and could depend on natural variation in their amount in a cell. Because HMGNs belong to a family of nucleosome-binding proteins, core and linker histones are expected among the proteins with the greatest abundance. We also noticed that there are no other distinct proteins or protein complexes that are significantly abundant.
Common for all samples are proteins with functions of epigenetic regulation and histone modification (see ES_N1, position #49—Jarid2; ES_N1, #37, and ES_N2, #120—Eed), cell cycle (ES_N1, #12—Aurkb; ES_N1, #79—Nsd2), transcriptional regulation (ES_N1, #44, and ES_N2, #173—H2ax, ES_N1, #34—Dmnt3a), chromatin remodeling (ES_N1, #21—Chd1; ES_N1, #49—Jarid2), DNA damage repair (ES_N1, #66—Mgmt; ES_N1, #59—Lig3). Some of the proteins are located in more than one list, including histone variant H2A.X. As expected, HMGN are detected among all binding partners (HMGN1: list ES_N1, #47, ES_N2, #192, and MEF_N1, #163; HMGN2: list ES_N2, #193, MEF_N1, #164).
To capture functional information on HMGN-binding partners, a comparative gene ontology (GO) Over-Representation Analysis (ORA) [1] was performed. ORA counts the number of proteins shared by an input set and each annotated set and applies a statistical test, such as the Fisher’s exact test, to calculate the statistical significance of the overlap between two sets. We first analyzed the protein sets—HMGN binding partners—using GO categories annotated by proteins that are related to various biological processes (PANTHER classification). A statistical significance threshold was selected as a conventional 5 × 10–2. HMGN binding partners from all four experiments (ES_N1, ES_N2, MEF_N1, and MEF_N2) yielded an over-representation of proteins from hundreds of various GO categories (Additional file 4: Table S2).
To reveal common GO categories, we overlapped these in a Venn diagram (Fig. 5A). We observed a prominent similarity between HMGN-specific and cell-specific preferred GO-categories (318 common categories vs. 184 unique ones), and 46 were found to be over-represented in all four experimental groups (Fig. 5A, black box). To focus on top GO categories preferred by HMGN binding partners, a set of all HMGN binding partners was subjected to the ORA test; 70 GO categories with an FDR-adjusted p value of < 0.05 (Additional file 4: Table S2, Common GO tab) were found. Next, we ranked top 20 GO categories by − log10 (P), where (P) is an FDA-adjusted p value (Fig. 5B). Proteins involved in chromatin and chromosome organization (GO:0051276; GO:0006325) showed the highest − log10 (P) value, i.e. they are over-represented among HMGN binding partners.
Fig. 5
Gene ontology analysis for the HMGN binding partners identified by chromatin proteomic profiling. A Venn diagram that shows the overlap between overrepresented GO categories (FDR > 5%) for HMGN1 and HMGN2 binding partners in both cell types. The black diamond shows the GO categories (46) in which proteins are overrepresented among both HMGNs and both cell types. B List of the top 20 out of 46 GO categories over-represented among both HMGNs and both cell types. C Cellular component GO-category domain analysis of HMGN1 and HMGN2 binding partners in both cell types. Top eight categories ranked by − log10 (P) value are depicted. Bar graphs − log 10 (P), P is FDR-corrected p value, blue and line graphs (enrichment score), red. D Molecular function GO category domain analysis of HMGN1 and HMGN2 binding partners in both cell types. Top five categories ranked by − log10 (P) value are shown
To gain additional insight into functional characteristics of HMGN binding partners, we conducted the ORA test using two other GO categories: cellular component (Fig. 5C) and molecular function (Fig. 5D). The GO term “chromatin” has the highest − log10 (P) value (over 11), and the highest enrichment score (over 14). In the “Molecular Function” GO analysis the terms of DNA-, chromatin- and nucleosome-binding proteins (enrichment score of over 40) are among the highest HMGN binding partners.
Next, we compiled a list of the identified HMGN binding partners in all four experimental groups, belonging to “Chromatin organization” and other selective GO categories (Additional file 5: Table S3, “Chromatin organization,” “Chromatin remodeling,” “Histone modification,, “DNA repair,” “DNA packaging,” “Chromatin assembly,” and other tabs). Each category contains numerous HMGN colocalizing partners. Several proteins (e.g., Smarca5, Mecp2, and Kat6b) were present in several categories. We concluded that, by co-localizing with numerous different factors, HMGN proteins contribute to modulation of chromatin architecture and function.
留言 (0)