
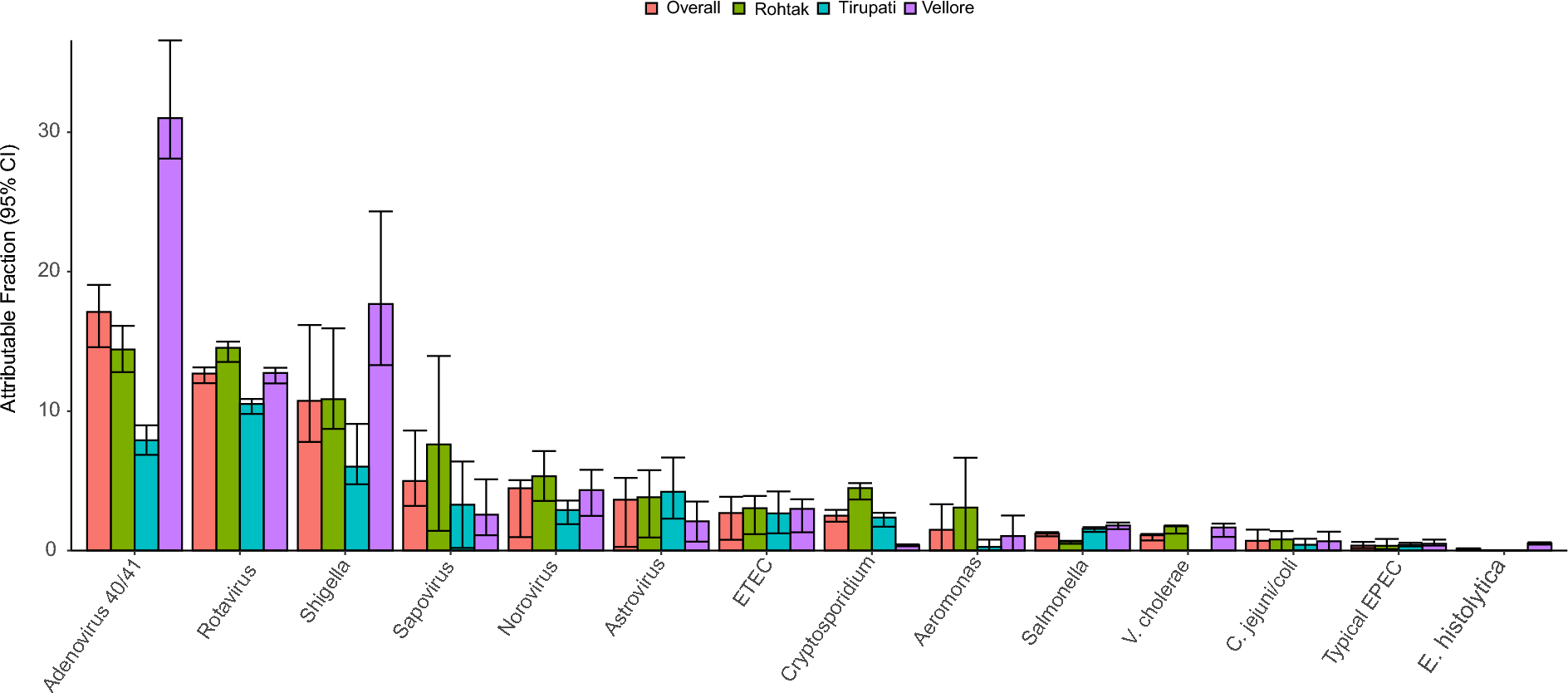
記住我


As the most suitable index for bacterial phylogeny and taxonomic identification, 16S rDNA was used to assess differences in gut bacteria community between the two groups. Results showed significant variations in bacteria composition from the phylum to species levels. The exploration of OTUs via UCLUST in QIIME software revealed that while both groups shared a large proportion of the OTUs (355 common OTU’s), IBD samples had 13 unique OTUs and the control had 32 unique OTUs (Fig. 1A). At the phylum level, the bacteria community structure of IBD patients had reduced levels of Firmicutes, Bacteroides, Fusobacteria, and Tenericutes but increased abundance of Proteobacteria and Actinobacteria compared with healthy controls. The top 10 abundant phyla between the healthy and IBD groups are presented in Fig. 1B and Table 1. We further explored the specific bacteria alterations between the two groups by examining the top 10 species of significant abundance, where increased abundance in IBD included Escherichia coli, Klebsiella pneumoniae, Bifidobacterium longum subsp. Longum, Bacteroides ovatus V975, and uncultured bacterium, while uncultured Bacteroides sp. and s_gut metagenome/human gut metagenome were reduced in abundance (Table 1, Fig. 1C). Community Heatmap map was used to intuitively express the size of the clustered data value at each classification level. The phylum-level clustering in IBD confirmed a significantly increased abundance of Proteobacteria, Actinobacteria, and Verrucomicrobia and a decreased abundance of 12 other phyla as sown in Fig. 1D. Moreover, group specific species classification tree revealed the changes at all levels (Fig. 1E). For instance, at the genus level, IBD samples had reduced Bacteroides, Dialister, Subdoligradulum, and Ruminococcus 2, but increased abundance of Escherichia-Shigella and Bifidobacterium.
Fig. 1
Gut bacteria community variations between IBD and healthy individuals. A Venn diagram; B Variation in the top 10 abundant phyla between groups; C Variation in the top 10 abundant species between the groups; D Community cluster heatmap at the phylum level; E Group specific species classification tree. N—Healthy control group; P—IBD group
Table 1 Species annotation of the top 10 gut flora with the largest abundance in each group at the phyla and species classification levelsAlpha- and beta-diversity changes in gut bacteria community in IBD patientsTo explore the differences in α-diversity index between the groups, four diversity indexes (Chao 1, ACE [abundance-based coverage estimator], goods coverage, and observed species) were used. These tools revealed significant differences in the bacteria diversity between IBD samples and normal controls by intuitively reflecting the median, dispersion, maximum, minimum, and abnormal values of species diversity in the groups. There was significantly reduced α-diversity in IBD samples compared to healthy controls; Chao 1(p = 0.009), ACE (p = 0.004), goods coverage (p = 0.021), observed specifications (p = 0.002) (Fig. 2A, B). To further confirm the difference between the two sample groups to the greatest extent, principal component analysis (PCA) and non-metric multidimensional scaling (NMDS) statistics were employed. PCA results showed more closely clustered IBD samples, indicating reduced α-diversity as compared to the more scattered healthy control samples, indicating a more diverse bacterial community composition (Fig. 2C). The NMDS statistical ranking method, as a nonlinear model, was used to overcome the shortcomings of the linear model (i.e., PCA) and better reflect the nonlinear structure of data. The multi-dimensional space generated by NMDS revealed the degree of difference between both the inter—and intra- groups (Fig. 2D). In the analysis of β-diversity index differences between the groups, the nonparametric test, Anosim, revealed a significant difference in β diversity between the two groups (Fig. 2E). The weighted UniFrac distance box chart further confirmed the increased β-diversity in the IBD group (p = 0.005) (Fig. 2F).
Fig. 2
α and β diversity variation in the groups. A Chao 1 box chart of α diversity differences between the groups; B Abundance-based coverage estimator box chart of α diversity differences between the groups; C PCA of the community composition of the groups; D NMDS analysis reflecting the nonlinear structure of the bacteria community composition of the groups; E Anosim group differences in β diversity; F Weighted UniFrac distance box chart of β diversity differences between the groups. N—Healthy control group; P—IBD group
Biomarker analysis and functional prediction between IBD and healthy controlThe observed differences were further analyzed to discover possible high-dimensional biomarkers and genomic features that differentiate IBD stool samples from normal controls using the LEfSe software. The results, including a cladogram, linear discriminant analysis (LDA) value distribution, and abundance comparison diagram of biomarkers with statistical differences between the two groups, revealed probable biomarkers for IBD. There was increased abundance and genomic features of the families Enterococcaceae and Lactobacillaceae and the genera Enterococcus, Lactobacillus, and Eggerthella, representing the microbial groups that play an important role in the IBD group, and serving as distinguishing biomarkers (Fig. 3A–C). Moreover, STAMP differential analysis revealed several bacteria communities that significantly differentiate the IBD group from the healthy group at the genus level, including reduced relative abundance of Dialister, Alistipes, Subdoligranulum, Ruminococcaceae UCG-002, UCG-005, UCG-010, and Coprococcus 2, but increased abundance of Anaerostipes, [Eubacterium] hallii group, and Eggerthella (Fig. 3D).
Fig. 3
Microbial biomarker analysis between IBD and healthy controls. A Cladogram of LEFSe analysis results in the IBD group; B LDA value distribution differentiating IBD group; C Relative abundance of the potential biomarker in the IBD group; D STAMP differential analysis of bacterial populations between the groups at the genus level
For functional prediction in IBD and its differential value, the PICRUSt software was used to infer the functional gene composition of samples by comparing the species composition information obtained from 16S sequencing data, to analyze the functional differences between the different groups and their value as biomarkers. Moreover, the COG homologous protein cluster and function classification database of prokaryotes was used to complement KEGG and reveal the functional composition of the flora more comprehensively. The KEGG and COG function prediction analyses of the metabolic function changes in the IBD group via STAMP analysis showed significantly increased factors such as carbohydrate metabolism and transport, transcription, xenobiotics biodegradation and metabolism, metabolism and transport of amino acids, and biosynthesis of other secondary metabolites (Fig. 4A, C), as associated with the heatmap analysis of the significant gene composition variations between the groups (Fig. 4B). There was also increased functional indication of immune system diseases and infectious diseases in the IBD group (Fig. 4A). LEfSe LDA analysis based on COG homologous protein cluster and function classification revealed significantly elevated carbohydrate transport and metabolism and RNA processing and modification in the IBD group as against reduced translation of ribosomal structures and biogenesis, and chromatin structure and dynamics (Fig. 4D). The abundance comparison of the increased functional items (as appeared in individual samples) in the two groups is further shown in Fig. 4E, F.
Fig. 4
Functional prediction and biomarker analysis of the groups. A KEGG STAMP analysis of the significant gene composition variations between the groups; B COG heatmap analysis of the significant gene composition variations between the groups; C COG STAMP analysis of the significant gene composition variations between the groups; D LDA value distribution and comparison of the abundance of functional items with statistical differences between the groups based on COG function prediction; E The comparison of abundance of RNA processing and modification function; F The comparison of abundance of carbohydrate metabolism and transport function
Variations in gut metabolomics between IBD and healthy controlsDifferential analysis of significant metabolitesA high-resolution nontargeted metabolomics analysis using ultra-high-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF MS) was carried out to identify metabolites, followed by strict checks and manual confirmation of results. The positive ion mode identified 2223 metabolites while the negative ion mode identified 1063 metabolites, yielding a combined total of 3146 metabolites. Further analysis revealed a total of 135 differential metabolites between IBD and healthy controls (Table 2). Based on univariate analysis (fold change (FC) analysis), all metabolites detected in positive and negative ion modes were screened for the differential metabolites (FC > 1.5- rose red, FC < 0.67- blue, p-value < 0.05) in a volcano plot. The significant differential metabolites were distributed among 33 classes and 14 superclasses of compounds (Fig. 5A, B). PCA and orthogonal partial least squares discriminant analysis (OPLS-DA) of both the negative and positive ion mode (Fig. 5C–F) along with their displacement test (Fig. 5G, H) confirmed a distinct set of differential metabolites associated with the groups.
Table 2 An overview of the metabolomic analysis outcomeFig. 5
Differential analysis of significant metabolites between IBD and healthy controls. A Volcano plot of significantly different metabolites according to molecular class in negative ion mode; B Volcano plot of significantly different metabolites according to molecular class in positive ion mode; C PCA score diagram of negative ion mode; D PCA score diagram of positive ion mode; E Negative ion mode OPLS-DA score plot; F Positive ion mode OPLS-DA score plot; G Negative ion mode OPLS-DA displacement test; H Positive ion mode OPLS-DA displacement test; I Multiple analysis of significant differences in metabolite expression in negative ion mode; J Multiple analysis of significant differences in metabolite expression in positive ion mode; K AUC of 6,7,4'-trihydroxyisoflavone; L AUC of [(2r,3 s,4 s,5r,6r)-3,4,5-trihydroxy-6-[2-(3-hydroxy-5-oxooxolan-3-yl)propoxy]oxan-2-yl]methyl (e)-3-(3,4-dihydroxyphenyl)prop-2-enoate (0.91). N—Healthy control group; P—IBD group
In further examination of the differential metabolites, the negative ion mode molecules revealed the top five upregulated differential metabolites as Calycosin, His-Met, 1,2,3-benzenetriol, G(8-o-4)fa sulfate, and.Alpha.-apooxytetracycline, with the top five downregulated being Lithocholic acid, Clausarin, Ginsenoside rh2, Isodeoxycholic acid, and Propylpyrazoletriol (Fig. 5I). The top five upregulated versus downregulated differential metabolites in the positive ion mode were P-methoxymethamphetamine, Apigenin, O-methylarmepavine, 2,5-dimethoxy-4-methylphenethylamine, and Luteolin, versus 1(2 h)-pyrimidineacetamide, n-[(1 s,3 s,4 s)-4-[[2-(2,6-dimethylphenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-4-hydroxy-.alpha.-(1-methylethyl)-2-oxo-, (.alpha.s)-, Isocaproic acid, Garcinolic acid, Anhydroecgonine methyl ester, and Salvinorin a, respectively (Fig. 5J). Furthermore, AUC (Area under the ROC Curve) analysis revealed several metabolites with high sensitivity and specificity in differentiating IBD from healthy individuals, including 6,7,4′-trihydroxyisoflavone (AUC = 0.92), thyroxine 4'-o-.beta.-d-glucuronide (AUC = 0.92), trichostachine (AUC = 0.91), normorphine (AUC = 0.90), and salvinorin a (AUC = 0.90). The top 20 metabolites in AUC measurement are presented in Table 2, while Fig. 5K, L shows representative diagrams of the AUC analysis.
Changes in metabolic pathways and function in IBDKEGG pathway enrichment analysis was carried out through the Fisher’s Exact Test to determine the significantly affected metabolic and signal transduction pathways in IBD. The results revealed altered metabolites (Fig. 6A, B) and 13 significantly affected pathways including vitamin digestion and absorption, primary bile acid biosynthesis, protein digestion, and absorption, thiamine metabolism, glutathione metabolism, ABC transporters, central carbon metabolism in cancer, and ferroptosis. The heatmap of differential metabolites in the largest pathway identified (ABC transport) is shown in Fig. 6C. Analysis of overall changes of KEGG metabolic pathway using differential abundance score and pathway enrichment is shown in Fig. 6D, E. Pathway hierarchy analysis showed that the changes in the IBD patients affected cancer function, cell growth and death, digestive system, lipid metabolism, membrane transport, and metabolism of cofactors, vitamins, and other amino acids (Fig. 6F). The specific metabolites dysregulated in these pathways are presented in Table 3. These results indicate significantly altered metabolomics and associated pathways in IBD patients compared to healthy individuals.
Fig. 6
Changes in metabolic pathways and function. A Negative ion pattern of significantly different metabolite hierarchical clustering heat map of individual samples within the groups; B Positive ion pattern of significantly different metabolite hierarchical clustering heat map of individual samples within the groups; C KEGG pathway differential metabolite clustering heat map of ABC transport; D KEGG metabolic pathway enrichment map (Bubble chart); E Differential abundance score maps for all differential metabolic pathways; F Differential abundance score map of all differential metabolic pathways (classified according to pathway hierarchy). N—Healthy control group; P—IBD group
Table 3 Dysregulated KEGG metabolic pathways and associated metabolites in IBDCorrelation of differential flora and metabolites in IBDTo further assess the metabolomics changes in the IBD group, the relative abundance of three flora of significant difference at the genus level (Eggerthella, Enterococcus, Lactobacillus) and 89 significantly differential metabolites were sorted and analyzed. Spearman analysis was used to generate a correlation coefficient matrix heat map and hierarchical clustering heat map (Fig. 7A, B) to reflect the similarities and differences of expression patterns of the significant flora and metabolites. There were 1144 pairs of significantly related differential bacteria and metabolites, of which 285 pairs had a more significant correlation (P < 0.01). The matrix not only showed the correlation between significantly different flora and metabolites but also between significantly different metabolites-metabolite and flora-flora. Enterococcus had positive significant correlation with 17 metabolites including cholic acid, calycosin, and N-nitrosopyrrolidine (p < 0.001), and flavin mononucleotide, apigenin, L-valine, and 3alpha,7beta,12alpha-trihydroxy-5beta-cholan-24-oic acid (p < 0.01), but negatively significant correlation with 43 metabolites including ginsenoside rh2, androsterone, indole-3-carboxaldehyde, salvinorin a, isodeoxycholic acid, and lithocholic acid (p < 0.001), and glycerol, uracil, oxypurinol, 25-hydroxycholesterol, glycolithocholic aid, xanthine, and hypoxyxanthine (p < 0.01). Eggerthella positively correlated with 13 metabolites including corydaline, delsoline, calycosin, apigenin, flavin mononucleotide, his-met, and luteolin (p < 0.01), but negatively correlated with 43 metabolites including hecogenin, salvinorin a, lithocholic acid, hypoxanthine, neomycin, Asiatic acid, piperonyl sulfoxide (p < 0.001). Lactobacillus had positive significant correlation with 9 metabolites including L-valine (p < 0.001), N-nitrosopyrrolidine, calycosin, apigenin, flavin mononucleotide, his-met, luteolin, and 1,2,3-benzenetriol (p < 0.05), but negative significant correlation with 11 metabolites including 25-hydroxycholesterol, androsterone, ginsenoside rh2, pristimerin, and cholesterol (p < 0.01) (Fig. 7B).
Fig. 7
Association analysis of flora and metabolites with significant difference between the groups. A Spearman correlation coefficient matrix heat map of significant difference flora and metabolites; B Spearman correlation analysis hierarchical clustering heat map of significant difference flora and metabolites. The correlation coefficient R is expressed in color, where R > 0 indicates a positive correlation and is represented by red, R < 0 indicates a negative correlation and is expressed in blue. The darker the color, the stronger the correlation. P-value reflects the significant level of correlation and was defined by P < 0.05 as *, P < 0.01 as * *, P < 0.001 as * * *; C Correlation network diagram. The color of the line represents the positive and negative value of the correlation coefficient between the two (blue represents negative correlation and red represents positive correlation), and the thickness of the line is directly proportional to the absolute value of the correlation coefficient. The node size is positively correlated with its degree, that is, the greater the degree, the larger the node size. Spearman correlation analysis network of significant difference flora and metabolites; D, E Representative scatter diagram of correlation
Moreover, the Cytoscape 3.5.1 software was used to generate a different perspective of the relationship between the flora and metabolites. The network chart revealed a total of 8 pairs of flora-metabolites with significant positive correlation and 44 pairs with a significant negative correlation that connect the three flora (Fig. 7C). The distribution characteristics of the correlation were also generated with a scatter diagram, which revealed 52 pairs of correlated flora-metabolites with significant levels. For example, the scatter diagram of the correlation between Eggerthella and piperonyl sulfoxide, and Enterococcus and N-nitrosopyrrolidine are shown in Figs. 7D, E. These observations do not only reveal changes in IBD but also provide important data in the search for therapeutic targets and diagnostic markers in IBD. However, more specific and detailed studies are required.
留言 (0)