Clinical samples
The frozen hippocampi of persons with AD dementia with APOE4/E4 carriers (N = 9) and no-cognitive impairment (NCI) with APOE3/E3 carriers (N = 7) were collected from the University of Southern California (USC) Alzheimer Disease Research Center (ADRC) Neuropathology core, which was approved by USC’s Institutional Review Board (IRB) protocol (HS-16-00888). The frozen inferior frontal lobe (Brodmann area 10) of the individuals with NCI and the APOE3/E3 carriers (N = 12) and APOE3/E4 carriers (N = 10) and persons with AD patients and the APOE3/E3 (N = 12) and APOE3/E4 genotypes (N = 10) were obtained from the Rush Alzheimer’s Disease Center (RADC) at the Rush University Medical Center. Rush Memory and Aging Project was approved by an Institutional Review Board (IRB) of Rush University Medical Center.
Animals
ApoE3-TR and ApoE4-TR mice were a generous gift from Dr. Patrick Sullivan. The endogenous mouse ApoE was replaced by either human APOE3 or APOE4, created by gene targeting, as described previously [58]. All experiments were performed on age-matched animals (8 months of age) and were approved by the USC Animal Care Committee. Every effort was made to reduce animal stress and to minimize animal usage. The mice were anesthetized with isoflurane and perfused with PBS. The brains were split in half for further analysis.
Cell cultures
Primary astrocytes were obtained from C57JB6, ApoE3-TR, and ApoE4-TR mice pups and cultured, as described previously [59]. Briefly, cerebral cortices from each 1 to 3 day-old neonatal mouse were dissected in ice-cold Hanks’ Balanced Salt Solution (HBSS) (Corning, 21–021-CV) and digested with 0.25% trypsin for 20 min at 37 °C. Trypsinization was stopped by the addition of a 2-fold volume of DMEM (Corning, 10–013) with 10% fetal bovine serum (FBS) (Omega Scientific, FB-12) and 1% antibiotic-antimycotic (Anti-anti) (Thermo Fisher, 15,240,062). The cells were dispersed into a single-cell level by repeated pipetting and filtered through 100 μm cell strainers (VWR, 10199–658). After filtering, cells were centrifuged for 5 min at 1000 rpm and resuspended in a culture medium supplemented with 10% FBS and antibiotics. Then, cells were seeded in a 75 cm2 flask and cultured at 37 °C in 5% CO2. The medium was changed on the next day and then replaced every 3 days. These mixed glia cultures reached confluence after 7–10 days. The cells were then shaken at 250 rpm for 16 h at 37 °C to remove microglia and oligodendrocyte progenitor cells. The remaining cells were harvested by digestion with trypsin. At this stage, the culture contained 95% astrocytes and was used for further experiments.
Immortalized mouse astrocytes derived from human ApoE3 and ApoE4 knock-in mice [60] were gifts from Dr. David Holtzman and grown in DMEM/F12 (Corning, MT10090CV) containing 10% FBS, 1 mM sodium pyruvate (Thermo Fisher, 11,360,070), 1 mM geneticin (Thermo Fisher, 10,131–035) and 1% anti-anti.
cPLA2 antibody validation
cPLA2 antibodies from Santa Cruz Biotech (sc-376,636, sc-376,618 and sc-454, 1:400) and Sigma (SAB4502200, 1:1000) were tested in astrocytes transfected with cPLA2 siRNA (15 nM) for 48 hours, then cells were lysed with RIPA buffer. After transfer and blocking, antibodies were added and incubated overnight at 4 °C. HRP-anti-mouse or HRP-anti-rabbit were used as secondary antibodies (Supplementary Figure 1 A). To test the phosphorylated cPLA2 antibody, astrocytes were treated with 100 μM ATP for 20 minutes. Cells were lysate with RIPA buffer with proteinase and phosphatase inhibitors. After centrifugation, the supernatants were collected and added with 4X sample buffer followed by boiling 5 minutes at 95 °C. Then 10 μL of lysates were loaded into 4–15% TGX gel (Bio-rad). After transferring and blocking, phospho-cPLA2 antibody (#53044, cell signaling technology) (1:1000) was added and incubated overnight at 4 °C. HRP-anti-rabbit was used as secondary antibody. The membrane was stripped with stripping buffer for 15 minutes at room temperature after imaging and blocked again for 1 hour with 5% milk. cPLA2 antibody (sc-376,618, Lot J1521, Santa Cruz Biotech) (1:400) was added and incubated 1 hour at room temperature. HRP-anti-mouse was used as secondary antibody (Supplementary Figure 1B). To test the cPLA2 antibody performance in human samples, astrocytes lysate (~ 2 μg total protein/lane) and human cortex lysate (10 μg total protein/lane) were loaded into 4–15% TGX gel (Bio-rad). After transferring and blocking, cPLA2 antibody (sc-376,618, Lot J1791, Santa Cruz Biotech) (1:400) was added and incubated overnight at 4 °C. HRP-anti-mouse was used as secondary antibody (Supplementary Figure 1C).
Cell lysate and brain homogenate preparation
The immortalized or primary astrocytes were lysed with 1x RIPA buffer (Cell Signaling Technology, CST 9806) containing protease inhibitor cocktail (Sigma, P8340) and phosphatase inhibitor cocktail (Sigma, P0044), followed by centrifugation at 14,000 gs for 10 min at 4 °C. The supernatant was collected for further analysis.
The mouse cerebral cortex, human hippocampus, and inferior frontal cortex were weighed, then RIPA buffer containing protease inhibitor cocktail and phosphatase inhibitor cocktail was added as 1:30 (w/v). The tissue was then homogenized using a 2 mL glass Dounce tissue grinder, followed by centrifugation with 14,000 gs for 10 min at 4 °C. The supernatant was collected, and the concentration was measured by BCA kit.
cPLA2 protein enrichment
To detect the phosphorylated cPLA2 in mouse cortex homogenates, cPLA2 protein was enriched by immunoprecipitation. For each mouse sample, 5 μg of cPLA2 antibody (Santa Cruz Biotechnology, sc-376,618) was conjugated to 50 μL Dynabeads Protein G (Thermo Scientific, 10003D) for 1 hr. at room temperature, then 500 μg total protein in 500 μL RIPA was added to the cPLA2-beads complex and incubated with rotation overnight at 4 °C. The beads were washed with 0.1% PBST 3 times by rotation for 5 min. After washing, 30 μL of 1x sample buffer (Bio-Rad, 1,610,747) was added to the beads and heated for 10 minutes at 100 °C. The supernatant was collected by magnetic force and used for the further Western-blot assay.
Western blot
The cell lysates, cortex homogenate, and enriched cPLA2 proteins were separated by 4–15% mini-precast protein gels (Bio-Rad, 4,561,086) under reducing conditions and then transferred onto nitrocellulose membranes (Bio-Rad, 1,704,270). After transfer, membranes were blocked with 5% fat-free milk (Bio-Rad, 1,706,404) in TBST for 1 h at room temperature, followed by overnight incubation with the primary antibody in 5% BSA at 4 °C. Then, the membranes were incubated with HRP conjugated secondary antibody for 1 h at room temperature. Chemiluminescent HRP substrate (Millipore, WBKLS0500) was used for detection. Fujifilm LAS-4000 imager system was used to capture images, and the densitometric quantification was done by Gel Quant NET software.
The following antibodies and dilution factors were used: cPLA2 antibody (Santa Cruz Biotechnology, sc-376,618) (1:200), phospho-cPLA2 (Ser505) antibody (CST, 53044) (1:1000), phospho-ERK1/2 antibody (CST, 4370) (1:1000), ERK1/2 antibody (CST, 4595) (1:1000), p38 antibody (CST, 9212) (1:1000), phospho-p38 antibody (CST, 4511) (1:1000), GFAP antibody (CST, 12389) (1:1000), Iba-1 antibody (GeneTex, GTX100042) (1:1000), iNOS antibody (CST, 13120) (1:1000), β-actin antibody (CST, 3700) (1:1000), β-tubulin antibody (CST, 2146) (1:1000), synaptophysin antibody (CST, 36406) (1:1000), Na,K-ATPase antibody (CST, 3010S) (1:1000), ApoE4 antibody (CST, 8941S) (1:1000), HRP-linked anti-mouse IgG (CST, 7076) (1:2000), HRP-linked anti-rabbit IgG (CST, 7074) (1:2000).
qPCR
The cells and brain specimens were harvested, and RNA was extracted using an RNA extraction kit (Thermo Fisher, K0731). Synthesis of cDNA was done using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 4368814). qPCR was performed using the PowerUp SYBR Green Master Mix (Thermo Fisher, A25742). The following primers were synthesized by Integrated DNA Technologies. The cPLA2 sense (5′-CTGCAAGGCCGAGTGACA-3′) and antisense (5′-TTCGCCCACTTCTCTGCAA-3′); mouse Tnfα sense (5′-GCCTCTTCTCATTCCTGCTTG-3′) and antisense (5′-CTGATGAGAGGGAGGCCATT-3′); mouse Il1β sense (5′-GCAACTGTTCCTGAACTCAACT-3′) and antisense (5′-ATCTTTTGGGGTCCGTCAACT-3′); mouse Il6 sense (5′-TAGTCCTTCCTACCCCAATTTCC-3′) and antisense (5′-TTGGTCCTTAGCCACTCCTTC-3′); mouse Ccl2 sense (5′-GTCCCTGTCATGCTTCTGG-3′) and antisense (5′-GCTCTCCAGCCTACTCATTG-3′); mouse Mip1α sense (5′- TGAAACCAGCAGCCTTTGCTC-3′) and antisense (5′-AGGCATTCAGTTCCAGGTCAGTG-3′); mouse Mip2 sense (5′-ATCCAGAGCTTGAGTGTGACGC-3′) and antisense (5′- AAGGCAAACTTTTTGACCGCC-3′); mouse β-actin sense (5′-ACCTTCTACAATGAGCTGCG-3′) and antisense (5′-CTGGATGGCTACGTACATGG-3′); human TNFα sense (5′-ACTTTGGAGTGATCGGCC-3′) and antisense (5′-GCTTGAGGGTTTGCTACAAC-3′); human IL1β sense (5′-ATGCACCTGTACGATCACTG-3′) and antisense (5′-ACAAAGGACATGGAGAACACC-3′);
human IL6 sense (5′-CCACTCACCTCTTCAGAACG-3′) and antisense (5′-CATCTTTGGAAGGTTCAGGTTG-3′); human CCL2 sense (5′- TGTCCCAAAGAAGCTGTGATC-3′) and antisense (5′-ATTCTTGGGTTGTGGAGTGAG-3′); human GAPDH sense (5′-ACATCGCTCAGACACCATG-3′) and antisense (5′-TGTAGTTGAGGTCAATGAAGGG-3′).
AA and DHA efflux assays
To investigate arachidonic acid (AA) and docosahexaenoic acid (DHA) release by cPLA2 and iPLA2 activation, respectively, we performed an AA and DHA efflux assay as described previously [2]. ApoE3 and ApoE4 primary astrocytes were seeded at 5000 cells/well in 96-well plates. After 24 h, the culture medium was changed with serum-free DMEM containing fatty acid-free BSA (5 mg/mL) (Sigma, A9647) and 3H-AA (1 μCi/mL) or 14C-DHA (1 μCi/mL) (Moravek) for 24 h. The cells were then washed twice with 100 μL of DMEM, and 100 μL of DMEM containing BSA (5 mg/mL) was added. After 30 minutes, the medium was removed, and 100 μL of ATP (100 μM) in DMEM without BSA was added. After 15 minutes, the cell culture medium was collected and transferred to scintillation vials filled with 3 mL of scintillation cocktail. The cells were solubilized in 90 μL of NaOH (0.5 N) for 5 minutes, neutralized with 60 μL PBS, and then transferred to scintillation vials filled with 3 mL scintillation cocktail. After rigorous mixing, the vials were counted in a Beckman LS6500 liquid scintillation counter (Beckman Coulter). The efflux of AA and DHA were assessed by the ratio of the corresponding fatty acid in the medium to total (medium and cell lysate). The change of AA and DHA efflux was calculated by subtracting the levels of AA and DHA in the ATP treated group to ATP non-treated group for each genotype. WT primary astrocytes were plated and labeled with 3H-AA (1 μCi/mL) or 14C-DHA (1 μCi/mL) as described above. Then, the cells were washed twice with 100 μL of DMEM. After wash, 10 μL of DMEM containing BSA and 0.2 μM recombinant ApoE3 or ApoE4 protein were added. After 24 h, the medium was removed, and 100 μL of ATP (100 μM) in DMEM without BSA was added. The AA and DHA efflux were measured as described above after 15 minutes.
cPLA2 activity assay
cPLA2 activity was detected by the cPLA2 activity assay kit (Cayman Chemical, 765,021). The mouse cortex was homogenized into HEPES buffer (50 mM, pH 7.4, containing 1 mM EDTA) as 1:10 (w/v), and the supernatant was collected after centrifuged and used for cPLA2 activity detection.
Immunoprecipitation
Immortalized ApoE4 astrocytes were cultured in a 100-mm dish for 18 hours and then were lysed with RIPA containing protease and phosphatase inhibitors. The lysates were used for immunoprecipitation with an anti-cPLA2 antibody or species-matched IgG. After elution, cPAL2 and p38 were detected by Western-blot.
p38 MAPK inhibition experiment
ApoE4 primary astrocytes were seeded in a 24-wells plate with the intensity of 100,000 cells per well. Forty-eight hours later, cells were pre-treated with p38 MAPK inhibitors – SB202190 (10 μM, Sigma, S7076) or SB203580 (10 μM, Sigma, S8307) in the DMEM culture medium without FBS for 20 minutes, followed by the treatment with vehicle or TNFα (10 ng/mL) (R&D Systems, 210-TA-005) plus IFNγ (100 ng/mL) (Sigma, SRP3058) together for 30 minutes. Then, the cells were lysed with RIPA. Total and phosphorylated cPLA2 and p38 were detected by western-blot.
LTB4 and PGE2 measurement
For the LTB4 and PGE2 measurements in the human brain samples, brain tissue was weighed, then PBS containing 1 mM EDTA, 10 μM indomethacin (Cox inhibitor, Sigma I8280), and 10 μM NDGA (Lox inhibitor, Sigma 479,975) as 1:10 (w/v) were added. The tissue was then homogenized using a 2 mL glass Dounce tissue grinder, followed by centrifugation with 8000 x g for 10 minutes at 4 °C. The supernatant was collected, and the protein concentration was measured using a BCA kit. LTB4 and PGE2 levels were detected by the assay kit (LTB4 ELISA Kit, Cayman Chemical, 10,009,292; PGE2 ELISA Kit, Cayman Chemical, 500,141).
For the LTB4 measurement in the cells, ApoE3 and ApoE4 primary astrocytes were seeded in a 24-wells plate with the intensity of 100,000 cells per well. Forty-eight hours later, cells were pre-treated with cPLA2 inhibitor-Pyrrophenone (500 nM, Sigma, 5,305,380,001) in the DMEM culture medium without FBS but containing N2 supplement for 30 minutes, followed by the treatment with vehicle or TNFα (10 ng/mL) (R&D Systems, 210-TA-005) plus IFNγ (100 ng/mL) (Sigma, SRP3058) together for 18 hours. Then, the culture media and cell lysate were collected. LTB4 levels were measured in a 4-fold concentrated medium using the assay kit.
ApoE4 primary astrocytes were seeded in a 24-wells plate with the intensity of 100,000 cells per well. Forty-eight hours later, cells were transfected with cPLA2 or non-target (NT) siRNA (10 nM) for 48 hours, followed by the treatment with vehicle or TNFα (10 ng/mL) plus IFNγ (100 ng/mL) together for 24 hours. Then, the culture media and cell lysate were collected. LTB4 levels were measured in a 4-fold concentrated medium by the assay kit.
ROS measurement
ROS were detected by the DCFDA cellular ROS detection assay kit (Abcam, ab113851). ApoE3 and ApoE4 primary astrocytes were seeded in dark, clear bottom 96-wells plate with the intensity of 20,000 cells per well. Forty-eight hours later, cells were pre-treated with cPLA2 inhibitor (1 μM) in the DMEM culture medium without FBS but containing N2 supplement for 30 minutes, followed by the treatment with vehicle or TNFα (10 ng/mL) plus IFNγ (100 ng/mL) together for 24 hours. After removing the media and washing plate once with 1x assay buffer, the cells were stained with DCFDA solution (100 μL/well) for 45 minutes at 37 °C in the dark. Then, the DCFDA solution was removed, and the 1x assay buffer (100 μL/well) was added to the plate. ROS levels were measured using a fluorescent plate reader at Excision/Emission = 485/585 nm.
Assessment of cellular distribution of cPLA2 in synaptosomes
Synaptosomes prepared from postmortem human frontal cortices using an established method with minor modification [61]. Briefly, the postmortem human frontal cortical tissue (about 100 mg) were homogenized in 10 volume (w:v) of ice-cold homogenization buffer (10 mM HEPES, pH 7.4, 0.32 M sucrose, 0.1 mM EDTA containing EDTA-free protease inhibitor cocktail (Roche, 04693159001) and 0.2% 2-mercaptoethanol) using a Teflon/glass homogenizer (10 strokes). The homogenates were cleared by centrifugation (1000 x g for 10 min), and the supernatants were centrifuged at 15,000 x g at 4 °C for 30 min to pellet the synaptosomes (P2 fraction). The synaptosomes were washed twice at 4 °C in 1 mL of ice-cold oxygenated KR (Kreb’s-Ringer) solution (25 mM HEPES, pH 7.4, 118 mM NaCl, 4.8 mM KCl, 25 mM NaHCO3, 1.3 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM glucose, 100 μM ascorbic acid, EDTA-free protease inhibitor cocktail). The synaptosomes were then resuspended in 1 mL of K-R solution, and the protein concentrations were determined by the BCA kit. Two hundred μg synaptosomes were incubated with 0.1 μM of Aβ42, rE3, rE4 or 0.7 μM Ceramide-1-phosphate (C-1-P) (Sigma, C4832) in 200 μL oxygenated KR buffer for 30 min at 37 °C followed by incubation with 2.5 μM C-1-P for 15 min. Upon completion of incubation, an ice-cold protein phosphatase inhibitor cocktail (Roche, 04906837001) was added and placed on ice for 5 min, and synaptosomes were pelleted by centrifugation.
The cytosolic and membranous fractions of the synaptosomes were isolated by centrifugation. The synaptosomes were briefly sonicated (Kontes Micro Cell Disrupter) in 500 μL of immunoprecipitation buffer (25 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM EDTA, protease and protein phosphatase inhibitor cocktails, and 0.02% 2-mercaptoethanol and centrifuged at 48,000 x g for 15 min at at 4 °C. The resultant supernatant was collected as the cytosolic fraction, and the pellet was dissolved in 500 μL immunoprecipitation buffer containing 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40 and incubated at 4 °C with end-to-end shaking for 1 h. Then, the pellet was centrifuged again at 48,000 x g for 15 min at 4 °C to collect the supernatant as the membrane fraction. cPLA2 were isolated by immunoprecipitation with 1 h incubation at 4 °C with anti-cPLA2 antibodies (Santa Cruz Biotechnology, sc-376,636 and sc-454) and followed by overnight incubation with Dynabeads Protein G (Thermo Scientific, 10004D). After three washes with 1 mL of ice-cold 0.1% TBST, 25 μL diluted (1.5x) sample buffer (Bio-rad, 1,610,747) was added to the beads and boiled for 5 min at 95 °C. cPLA2 protein expression was determined by western-blot imaging with anti-cPLA2 (Santa Cruz Biotechnology, sc-376,618) antibody. Beta-actin and Na,K ATPase were used as markers for loading control.
LDH assay in synaptosomes
To assess synaptosomes membrane integrity, we performed a LDH activity test (Thermo Fisher, C20300) [28]. The synaptosomes were isolated and incubated for 1 hour at different conditions. Then, 50 μL of synaptosome in KR buffer with 1% Triton X-100 (total LDH) or without Triton X-100 (free LDH) were incubated with 50 μL LDH reaction mixture for 30 minutes. After adding the stop solution, the absorbance at 490 nm and 680 nm were measured. The LDH activity was indicated as the absorbance at 490–680 nm.
Statistical analysis
Descriptive results are presented as the mean ± SEM. Data were analyzed using two-tailed Student’s t-test or two-way ANOVA. Statistical significance was present at p < 0.05.

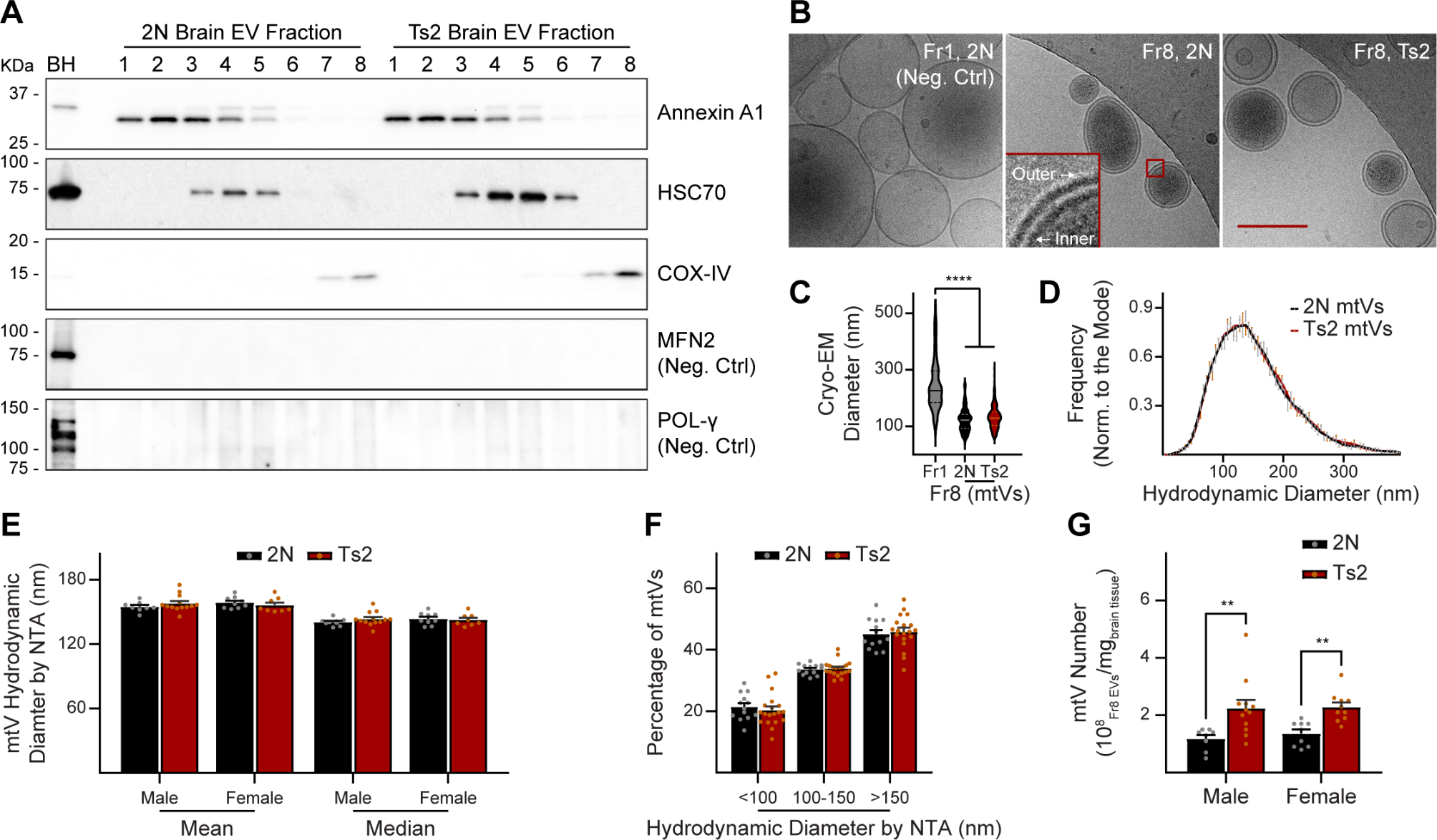

留言 (0)