Preparation of CSE
100% CSE was prepared by allowing the smoke generated by two 3R4F standard research cigarettes (Tobacco and Health Research Institute, University of Kentucky) with filter removed to flow directly into RPMI-1640 (sigma) in a glass flask with side flow smoke exposure system. Each cigarette burns for 8 min. The absorbance value of the collected 100% CSE was measured at the wavelength of 260 nm for inter batch quality control. The qualified 100% CSE was filtered and sterilized with 0.22 μm filter for the experiment.
Cell culture
Studies were performed using human bronchial epithelioid cell line 16HBE cells purchased from American type culture collection (ATCC). Cells were maintained in RPMI-1640 containing 10% Fetal Bovine Serum (FBS; Gibco), 0.1kU/ mL penicillin and 0.1 mg/ml streptomycin (Beyotime Biotechnology) at 37 °C in a humidified 5% CO2 atmosphere. The passage number of all cells used has no more than 10.
Cell transfection
FABP4-specific short hairpin RNAs (shRNA; shRNA-FABP4-1, shRNA-FABP4-2) and nontargeting shRNA (shRNA-NC) were synthesized and purified by RiboBio. Briefly, 5 × 105 16HBE cells were transfected with 25 pmol shRNA using Lipofectamine® RNAiMAX (Invitrogen) according to manufacturer’s recommendation.
Sequences are as follows: shRNA-FABP4-1, sense 5’- GGATGTGATCACCATTAAA-3’, antisense 5’- TTTAATGGTGATCACATCC-3’; shRNA-FABP4-2, sense 5’- GGGATGTGATCACCATTAA-3’, antisense 5’- TTAATGGTGATCACATCCC-3’; shRNA-NC, sense 5’-GATCCCCCTTCTCCGAACG-3’, antisense 5’-AGCTAAAAATTCTCCGAAC-3’.
RNA extraction, complementary DNA (cDNA) synthesis, real‐time quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR)
Total cellular RNA was isolated from 5 × 106 16HBE cells using the Total RNA Isolation Kit (Biomarker), and 5 µg of total RNA was used to reverse into cDNA using the PrimeScript™ RT reagent kit (Takara), according to manufacturers’ recommendations. Subsequently, the qRT‐PCR were performed using One Step TB Green® PrimeScript™ PLUS RT-PCR Kit (Takara) and detected by StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific).
Primer sequences were as follows: for human FABP4, forward 5’-ATGGGGGTGTCCTGGTACAT-3’, reverse 5’-CTTTCATGACGCATTCCACCA-3’; for human GAPDH, forward 5’-CACTAGGCGCTCACTGTTCT-3’, reverse 5’-GCCCAATACGACCAAATCCGT-3’.
Western blot (WB) analysis
Total cellular protein was prepared from 1 × 107 16HBE cells using 1 ml CelLytic™ MT Cell Lysis Reagent (Sigma) containing Phosphatase Inhibitor Cocktail (abcam). The proteins (20 µg/lane) were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto 0.45 µm polyvinylidene fluoride (PVDF) membranes (Millipore), and blocked with Tris-buffered saline containing 1% Tween-20 (TBST, pH 7.6) and 5% skimmed milk powder at room temperature for 1 h. After washed with TBST, the blots were incubated with primary antibodies at 4 °C overnight. After additional three more washes with PBS (15 min each time), the corresponding horseradish peroxidase (HRP)-linked secondary antibodies were used at room temperature for another 1 h. Finally, the protein bands were visualized with Chemiluminescent Western Blot Reagents (Thermo Fisher Scientific) and scanned with the PDQuest 7.2.0 software (Bio-Rad). Protein expression was quantified using ImageJ software (version 1.8.0; National Institutes of Health).
Primary antibodies: rabbit anti-FABP4 (1:1,000, 2120S, Cell Signaling technology); rabbit anti-GAPDH (1:1,000, 5174S, Cell Signaling technology); rabbit anti-Cox-2 (1:1,000, 12282S, Cell Signaling technology); rabbit anti-iNOS (1:1,000, 13120S, Cell Signaling technology); rabbit anti-p38 (1:1,000, 8690S, Cell Signaling technology); rabbit anti-p-p38 (1:1,000, 4511S, Cell Signaling technology); rabbit anti-p-MK2 (1:1,000, 3041S, Cell Signaling technology).
HRP-linked secondary antibody: Anti-rabbit IgG, HRP-linked Antibody (1:3,000, 7074S, Cell Signaling technology).
Cell Counting Kit-8 (CCK-8) assay
1 × 104 16HBE cells/well were seeded into 96-well plates and transfected with corresponding shRNA at 37 °C for 24 h. After 1 h treatment with or without 30 μM asiatic acid, an agonist of p38 MAPK at 37 °C, 100 µl fresh RPMI-1640 completement medium containing 2% CSE were replaced and cultured for additional 48 h. Subsequently, Cell Counting Kit-8 (MedChem Express) was used to determine the cell viability according to the manufacturer’s recommendation. The normal 16HBE cells without transfection and treatment were used as control.
Tunel assay
5 × 104 16HBE cells/well were seeded into 96-well plates and transfected with corresponding shRNA at 37 °C for 24 h. After 1 h treatment with or without 30 μM p38 agonist asiatic acid at 37 °C, 500 µl fresh RPMI-1640 completement medium containing 2% CSE were replaced and cultured for additional 48 h. Subsequently, One Step TUNEL Apoptosis Assay Kit (Beyotime Biotechnology) was used to determine the cell apoptosis according to the manufacturer’s recommendation. The normal 16HBE cells without transfection and treatment were used as control. The percentage of apoptotic cells was calculated using ImageJ software (version 1.8.0; National Institutes of Health).
Elisa assay
2 × 105 16HBE cells were seeded into cell culture dishes and transfected with corresponding shRNA at 37 °C for 24 h. After 1 h treatment with or without 30 μM asiatic acid, an agonist of p38 MAPK at 37 °C, 8 ml fresh RPMI-1640 completement medium containing 2% CSE were replaced and cultured for additional 48 h. Subsequently, Human TNF alpha SimpleStep ELISA® Kit, Human IL-1 beta SimpleStep ELISA® Kit, Human IL-6 SimpleStep ELISA® Kit (abcam) were used to determine the levels of inflammatory cytokines in culture supernatant according to the manufacturer’s commendation. The normal 16HBE cells without transfection and treatment were used as control.
Measurement of oxidative stress
Production of ROS and MDA, and SOD activity in 16HBE cells was measured by Cellular ROS Detection Assay Kit (ab186029; Abcam), Lipid Peroxidation (MDA) Assay Kit (ab118970; Abcam) and SOD Activity Assay Kit (ab65354; Abcam), respectively, strictly according to the manufactures’ protocols. Briefly, control or transfected 16HBE cells were treated with or without 30 μM asiatic acid for 1 h, followed by being cultured in fresh RPMI-1640 medium containing 2% CSE for additional 48 h. Subsequently, about 2 × 106 cells were harvested for corresponding Assay Kits. All experiments were performed in triplicate and data were shown as relative level or activity after normalization to control group.
Statistics
All data were shown as the mean ± standard deviation from experiments in triplicate. Comparisons between multiple groups were evaluated using one-way ANOVA followed by Tukey’s test using GraphPad Prism 8. P value < 0.05 was considered significant.

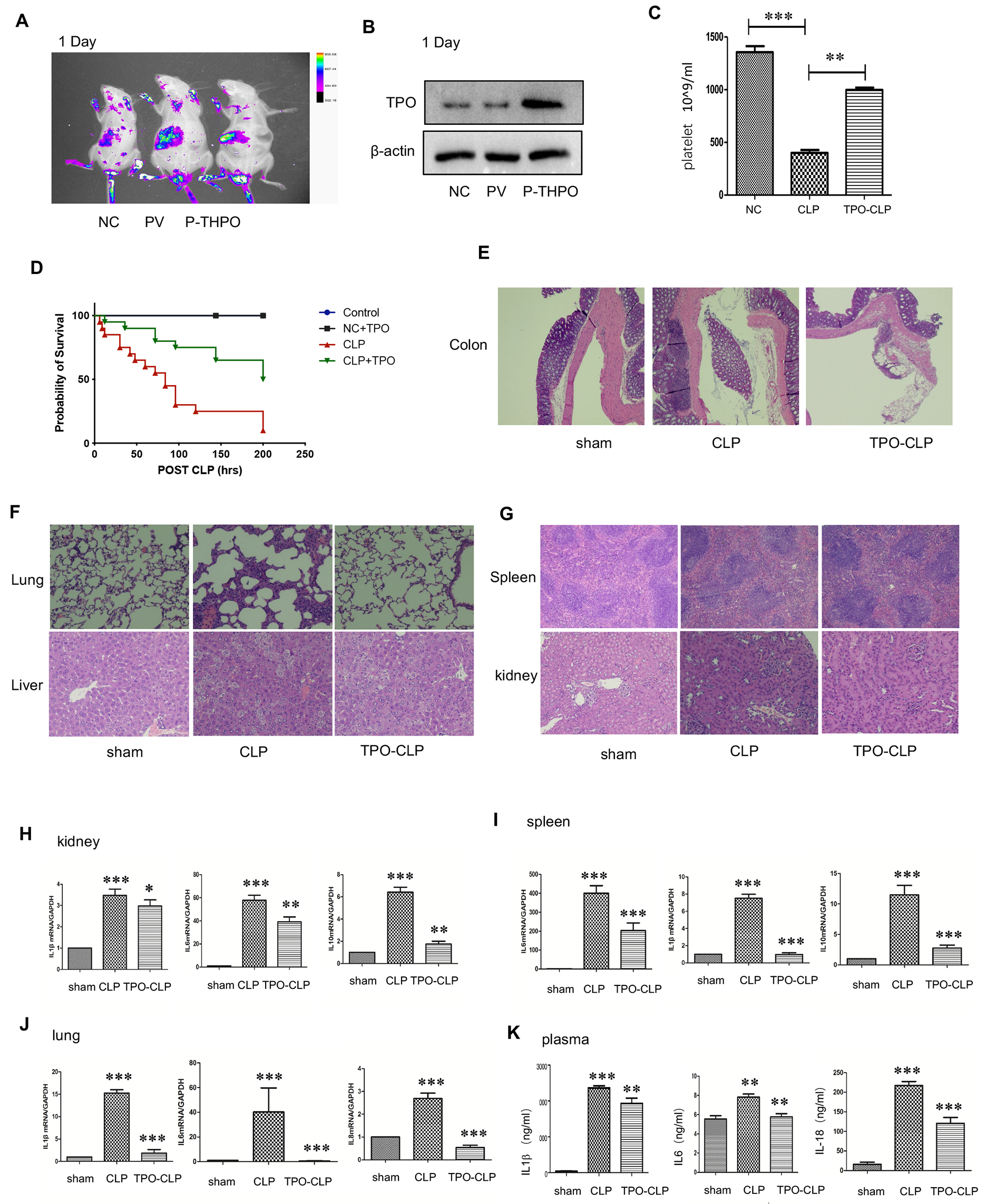

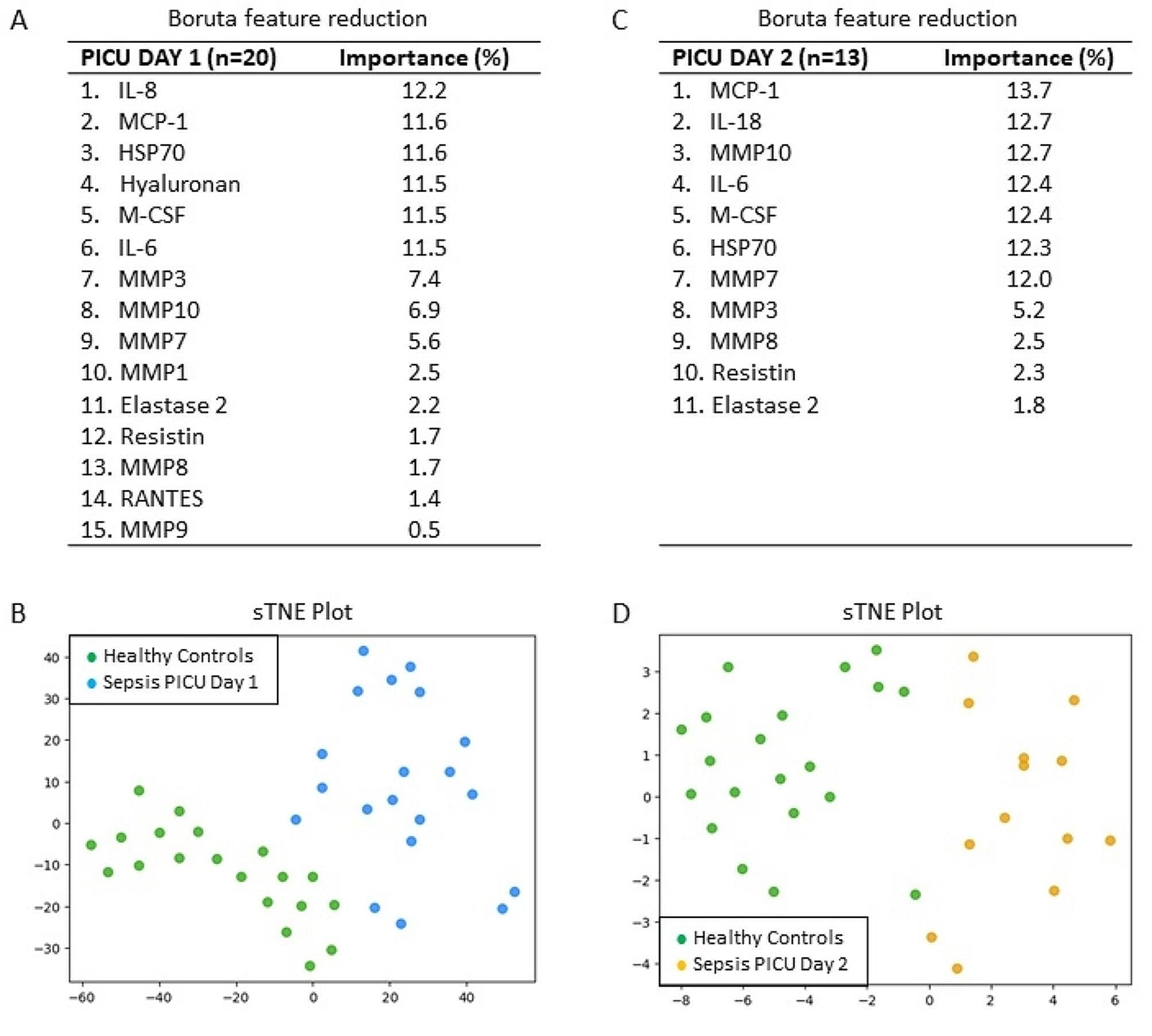
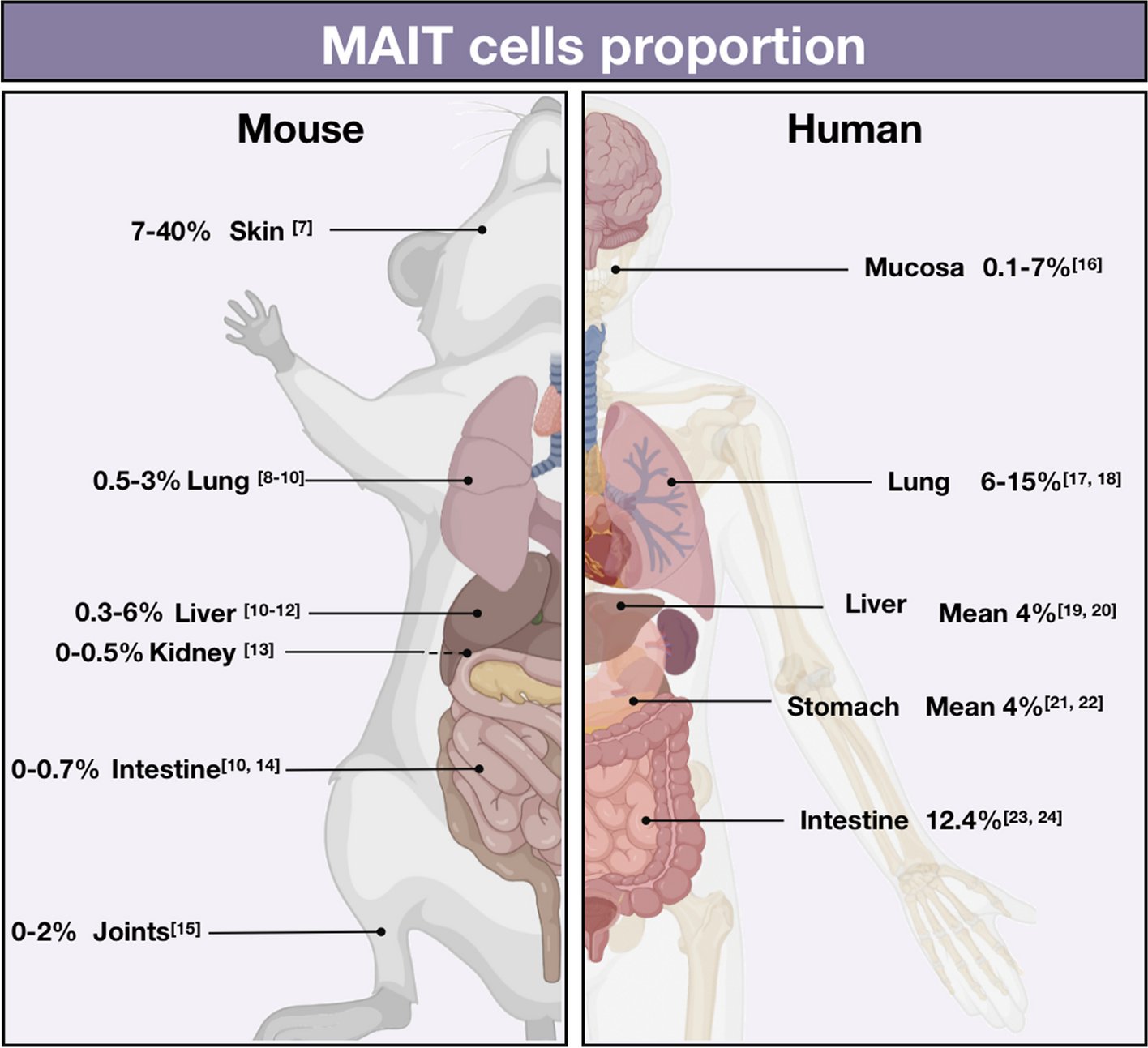
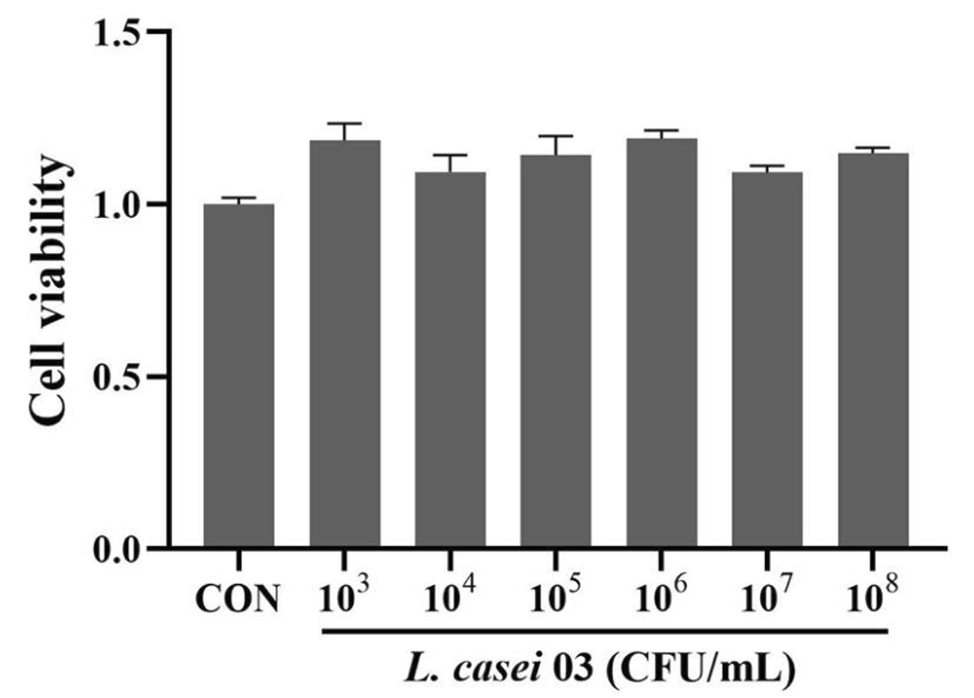

留言 (0)