
記住我


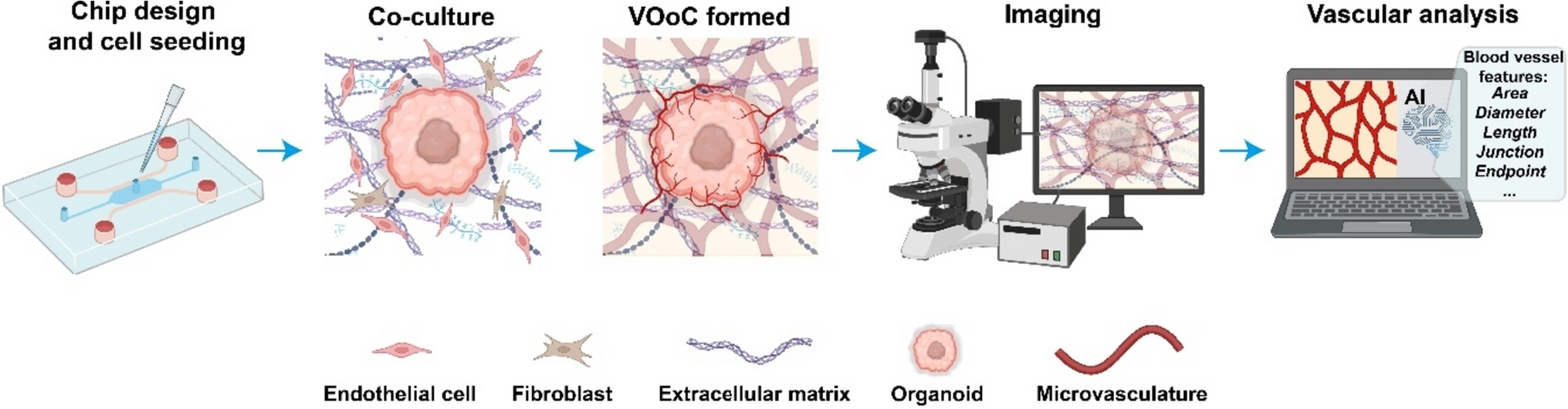
Human primary RFP-labeled HUVECs (Alphabioregen, #RFP4) were cultured in T75 flasks (Corning, #734–2705) in MV2 (PromoCell, #C-22121) supplemented with 1% penicillin/streptomycin (Sigma, #P4333) and used at passage 4 till passage 8 for all experiments. Before use, a T75 flask was pre-coated with 50 µg/mL Purecol (Advanced BioMetrix, #5005-B) for 30 min at 37 °C in PBS (phosphate-buffered saline, Life tech #20,012,068) and then washed twice with sterile PBS. Cryopreserved Upcyte® Human Hepatocytes (UHH) from donor 653–03 were purchased from Upcyte technologies GmbH (Hamburg, Germany) and cultured as previously described [38]. Briefly, UHH were thawed in Thawing Medium (Upcyte technologies, #MHE001) seeded at a density of 10,000 cells/cm2 in collagen-type I-coated T75 flasks (Thermo, #156,499) and cultured in High-Performance Medium (HPM, Upcyte technologies, #MHE003). Media were replaced three times a week. All cells regularly tested negative for mycoplasma. For all cells, culture was performed in a humidified incubator at 37 °C and 5% CO2.
Spheroids and organoids generationTo form hepatic spheroids, UHH at 100% confluency was dissociated using TrypLE™ Express Enzyme (Thermo, #12,604,021), and viability was assessed using the trypan blue dye exclusion method. 100 µl of HPM containing 20,000 UHH cells was transferred into each well of a Costar® ultra-low attachment 96-well plate (Corning, #CLS3474-24EA) and centrifuged for 1 min at 100 × g. For pre-vascularized spheroids, a mix of 1000 RFP-labeled HUVECs and 20,000 UHH were used. The plate was then placed in a humidified incubator (37 °C and 5% CO2). After 48 h, the spheroids were transferred onto the OrganoPlate Graft. Human hepatic organoids were generated from human fetal tissue as previously described [39]. After that, hepatic organoids were dissociated to single cells, and 6000 hepatocytes were added to each well of a Costar® ultra-low attachment 96-well plate in 100 µl of human Hep-Medium [39] and centrifuged for 5 min at 100 × g. After 3 days, organoids were transferred to the OrganoPlate Graft.
Microfluidic cell cultureOrganoPlate culture was performed using the OrganoPlate Graft with 400 µm × 220 µm (w × h) channels (Mimetas BV, the Netherlands). Phaseguides had dimensions of 100 µm × 55 µm (w × h). Gel and perfusion channels had a length of 9 mm and 13 mm, respectively, with a downstream phaseguide wall interface of approximately 132 and 164 degrees. The phaseguide stability is expected to be around 200 Pa [40]. 2.5 µl of gel composed of 4 mg/ml Collagen I (AMSbio Cultrex 3D Collagen I Rat Tail, 5 mg/ml, #3447–020-01), 100 mM HEPES (Life Technologies, #15,630–122), and 3.7 mg/ml NaHCO3 (Sigma, #S5761) was dispensed in the gel inlet and incubated 15 min at 37 °C. Endothelial cells were detached by use of TrypLE™ Express Enzyme (Thermo, #12,604,021), counted and pelleted (5 min, 300 × g). The cells were applied to the system by seeding 2 µl of 1 × 107 of cells/ml in the inlets of the perfusion channels. Subsequently, 50 µl of MV2 medium was added to the same inlets and the OrganoPlate was incubated in humified conditions at 37 °C and 5% CO2 for at least 1 h until cells attached on the bottom of the perfusion channels. After incubation, 50 µl medium was added to both channel outlets. The OrganoPlate was placed in the incubator (37 °C and 5% CO2) on an interval rocker switching between a + 14° and − 14° inclination every 8 min (OrganoFlow S, Mimetas) allowing bi-directional flow. Medium (50 µl each on inlet and outlet) was refreshed every 2–3 days. Endothelial tubes were cultured for 2–3 days before inducing microvascular bed formation.
Microvascular bed formationSprouting of RFP-labeled HUVEC was induced by addition to the graft chambers of 50 µl of MV2 medium containing angiogenic factors: 50 ng/ml vascular endothelial growth factor (VEGF), 20 ng/ml basic fibroblast growth factor (bFGF), 2 ng/ml Phorbol 12-myristate 13-acetate (PMA), and 500 nM Sphingosine 1-phosphate (S1P). Stock solutions were prepared as follows: 100 μg/ml murine VEGF in MilliQ water (Preprotech, #100–20), 50 µg/ml bFGF in MilliQ water (Peprotech, #G00832 100-18B), 1 mM S1P (Sigma, #G00918) in 5% 1 M HCl, 95% DMSO, 10 μg/ml PMA (Sigma, # P1585) in 1% DMSO, and 100 μg/ml. Sprouting mix was refreshed every 2–3 days.
Microvessels characterizationTo measure microvessels area and orientation, the microvascular bed was stained with Calcein AM green (Life Technologies, #C3099, 1:2000) in MV2 medium. After incubation for 30 min in the incubator (37 °C and 5% CO2) on an interval rocker switching between a + 14° and − 14° inclination every 8 min (OrganoFlow S, Mimetas), maximum intensity projections were acquired using the Micro XLS-C High Content Imaging Systems (Molecular Devices, US) at 4 × magnification. Maximum intensity projections from each chip were used to calculate the average microvessel orientation and microvessel area using the ‘orientationJ analysis’ tool and a modified version of the ‘morphology’ plugin in FIJI v.1.52. Data underlying Fig. 1f, e were obtained making use of automated image analysis routines that are described in supplementary Fig. 1c.
Fig. 1
The OrganoPlate Graft allows for the generation of robust microvascular beds. a Top and bottom views of the OrganoPlate Graft with 64 microfluidic units positioned underneath a 384 microtiter plate. Each microfluidic unit makes use of a 2 × 3 array of wells from the microtiter plate (insert image). b Sequence of steps for generating a microvascular bed. Step 1: the graft chamber is filled with Collagen I gel (depicted in blue) through the gel inlet A2. Step 2: endothelial cells are seeded against collagen I gel (depicted in red) in the two lateral perfusion channels and form tubules upon application of perfusion flow. Step 3: Angiogenic factors are added to the graft chamber B2 to induce sprouting of the lateral vessels and formation of the vascular bed. Step 4: once microvessels have reached the opening on the graft chamber, a target microtissue is positioned on top of the microvascular bed to initiate interaction. c Images of an RFP-labeled HUVEC vascular bed formation prior to application of angiogenic factors (left) and after (middle). The target tissue is then positioned on the center of the graft chamber opening (right). Scale bar: 200 µm. d Maximum intensity projection (i) and cross sections (ii, iii) of a microvascular bed stained against CD31 (green) and nuclei (blue). Microvessels with a lumen are apparent. Location of cross sections is indicated by dash lines. Scale bars: 200 µm. e Quantification of sprout area coverage in 64 microfluidic units per OrganoPlate for three different plates (n = 192) after microvascular bed formation. Significance was calculated using one-way ANOVA and shown as non-significant (ns, P > 0.05). f Average distribution frequency of the orientation of microvascular structures (90 degrees indicates horizontal alignment) from 3 different OrganoPlate Grafts. g Evaluation of manual and robotic placement accuracy of target tissue on top of the microvascular bed. Statistical significance was attributed to values of P < 0.05 as determined by unpaired Student’s t test
Hepatic microtissue culture with microvesselsAfter 4–5 days of endothelial cell sprouting, hepatic microtissues were placed in the circular opening in the graft chamber on top of the microvascular bed. Before microtissue placement, medium was refreshed as follows: for hepatic spheroids, medium was aspirated from all graft chambers, in- and outlets and replaced with 50 µl MV2 (in- and outlets) and 50 µl HPM (graft chamber); for hepatic organoids, medium was aspirated from all graft chambers, in- and outlets and replaced with 50 µl Hep-Medium (in- and outlets). Graft chamber was left dry to allow overlay of organoids with Matrigel.
For manual placement of hepatic spheroids, microtissues were picked up from the ultra-low attachment plate together with 20 µl culture medium using wide-bore tips (Pure™ 200G Pipet Tips, VWR, #53,225–682). The pipette tip was then placed above the circular opening of the graft chamber allowing the spheroid to slowly sediment from the pipette tip onto the microvascular bed. For placement with liquid handler, the same procedure was followed using the OT-2 robot (Opentrons). Position accuracy of the spheroids for both manual and automated placement was determined by measuring the relative position along XY axis versus the center of the graft chamber using FIJI v.1.52 and translating to a radial offset. The OrganoPlates were then placed in the incubator (37 °C and 5% CO2) and were kept static for 1 h to allow the spheroids to adhere to the underlying collagen-I matrix. This prevents displacement during initial stages of rocking as well as during subsequent medium changes.
Hepatic organoids were transferred manually using a standard p10 micropipette into the circular opening in the graft chamber. After placement, 5 µl Matrigel GFR (Corning, #356,231) was added on top of each organoid, and the gel was allowed to polymerize for 10 min at 37 °C. At the end of the incubation, 50 µl Hep-Medium was added to all graft chambers.
The OrganoPlates were then placed on an interval rocker switching between a + 14° and − 14° inclination every 8 min (OrganoFlow S, Mimetas) allowing bi-directional flow. Media were refreshed every 2–3 days.
Quantification of vessel intensity over timeFluorescent images were acquired every 2–3 days using an ImageXpress XLS Micro High content imaging system at 37 °C (4 × objective). FIJI v.1.52 was used to quantify the relative RFP-HUVEC vessel intensity by drawing a rectangular selection excluding the lateral perfusion channels and measuring the mean signal intensity.
Barrier integrity assayBarrier integrity of lateral vessels was examined by addition of MV2 media containing 0.5 mg/ml FITC-dextran (150 kDa, Sigma #46,946) to both perfusion channels (40 µl in the inlets and 20 µl in the outlets). 20 µl of fresh media without dye was added to the graft chamber. Images were acquired every 15 s for 8 min using an ImageXpress XLS Micro High content imaging system at 37 °C (4 × objective).
ImmunohistochemistryFor direct immunostaining in the OrganoPlate Graft of co-cultures, cells were fixed with 3.7% formaldehyde (Sigma, #252,549) in PBS (phosphate-buffered saline, Life tech #20,012,068) for 15 min. After washing twice for 5 min with PBS, permeabilization and blocking were carried out together using 0.3% Triton X-100 (Sigma, #T8787), 2% FCS, 2% bovine serum albumin (BSA) (Sigma, #A2153), and 0.1% Tween 20 (Sigma, #P9416) in PBS for 2 h. Subsequently, cells were incubated with primary antibodies overnight, washed three times, incubated with secondary antibodies for 2 h, and washed three times with 4% FCS in PBS. The following antibodies were used for immunohistochemistry: Mouse a-CD31 (Dako, M0823, Clone JC70A, 1:20), Goat a-Albumin FITC conjugated (Bethyl, A80-229F, 1:200), Mouse a-HNF4A (Thermo Fisher, MA1199, 1:500), Mouse a-MRP2 (Abcam, M2 III-6, ab3373, 1:100), Mouse a-acetylated tubulin (Sigma, T6793, 1:4000) Mouse a-E-cadherin (BD, 610,181, 1:300), and Goat a-Mouse AlexaFluor 647 (Thermo Fisher, #A31571, 1:250). After nuclear stain with Hoechst (Thermo Fisher Scientific, #H3570, 1:2000 in PBS) cells were stored in PBS (50 µl in all graft chambers and in- and outlets). Tissue clearing was performed using CUBIC reagents (TCI Chemicals, Cubic-L, and CUBIC-R +) according to manufacturer instructions.
All steps were performed at room temperature (RT) and under the presence of flow. Cells were imaged with the EVOS FL2 Auto, ImageXpress Micro XLS, and Micro XLS-C High Content Imaging Systems (Molecular Devices, US).
For immunostaining of hepatic spheroids sections, vascularized spheroids were first removed from the graft chamber at different timepoints of the co-culture, transferred to cryomolds, overlaid with Cryo-Gel (Leica, #39,475,237) and frozen by floating in an ethanol/dry ice bath. After solidification, cryomolds were stored at − 80 °C until cryosectioning. Cryosectioning was performed using a cryostat (Cryotome® FE, Thermo Scientific) at − 20 °C. 10 μm cryosections were collected on SuperFrost® Plus slides (Fisher Scientific, #10,149,870). The slides were air-dried and fixed with 3.7% formaldehyde (Sigma, #252,549) in PBS for 15 min. After washing twice for 5 min with PBS, sections were permeabilized with 0.5% Tween in PBS for 10 min and blocked with 2% BSA and 0.05% Tween in PBS. Sections were incubated with Mouse a-CD31 (Dako, M0823, Clone JC70A, 1:20), Goat a-Albumin FITC conjugated (Bethyl, A80-229F, 1:200) for 2 h, washed three times 5 min with washing solution (0.05% Tween in PBS) and incubated with secondary antibody for 1 h. After washing three times, nuclei were stained with DAPI (Thermo Fisher Scientific, #R37606). Sections were mounted with Vectashield (Vector Laboratories, #H-1400) and imaged with EVOS FL2 Auto.
Perfusion assayInterconnection and perfusion between the left and right side of the vascular network in the presence of microtissues were determined by addition of 100 µl MV2 medium containing 0.25 mg/ml FITC-Dextran (150 kDa, Sigma #46,946) to the left perfusion inlet, while 50 µl of MV2 medium was added to the left perfusion outlet and right in- and outlets. Medium in the Perfusion channels was removed for the course of the assay. Images were acquired every 60 s for 5 min using an ImageXpress XLS Micro High content imaging system at 37 °C (4 × objective). Perfusability of the vascular network was identified, when perfusion with fluorescent dye was observed via the lumen of vascular network from the left channel to the right perfusion channel. Perfusable chips were identified manually, when apparent flow of the dye was observed to traverse the vascular network reaching the right perfusion channel.
Transmission electron microscopyA co-culture of hepatic organoids with vascular bed was fixed with 1.6% glutaraldehyde for 24 h at 4 °C and removed from the OrganoPlate. Then, samples were kept in wash buffer (0.1 M cacodylate) until further processing. Samples underwent an additional fixation step with 1% osmium tetroxide and 1.5% potassium ferricyanide in wash buffer for 1 h in the dark at 4 °C. Further, the samples were progressively dehydrated in ethanol 70–100% and infiltrated with Epon resin for 2 days. Then, samples were embedded in Epon resin, which polymerized for 2 days at 60 °C. Ultrathin sections were cut using an ultramicrotome (Leica Ultracut UCT) and mounted on Formvar-coated copper grids. Sections were stained with 2% uranyl acetate in water and lead citrate. Sections were imaged using a Tecnai T12 electron microscope and an Eagle 4 k*4 k CCD camera (ThermoFisher Scientific).
Bile canaliculi stainingStaining with CDFDA was performed to visualize active bile canaliculi in the hepatic organoids co-cultured with microvessels. Briefly, a stock solution containing 5 mM 5-(and-6)-carboxy-2′,7′-dichloro-fluorescein diacetate (CDFDA) (Sigma-Aldrich, #2188) was prepared in 100% Dimethyl Sulfoxide (DMSO) (Sigma, #D8418), aliquoted, and stored at -20 °C. For the assay, culturing media were aspirated in all graft chambers and perfusion in- and outlets and 50 µL of Hep-Medium containing 5 µM CDFDA (1:1000), and Hoechst (Thermo Fisher Scientific, #H3570, 1:2000 in PBS) was added to each well. The OrganoPlate Graft was incubated for 30 min in the incubator (37 °C, 5%, CO2) on an interval rocker switching between a + 14° and − 14° inclination every 8 min (OrganoFlow S, Mimetas). As negative control, Hep-Medium media containing 0.1% DMSO and Hoechst (Thermo Fisher Scientific, #H3570, 1:2000 in PBS) was prepared. At the end of the incubation, chips were washed 6 times with PBS and imaged Micro XLS-C High Content Imaging Systems (Molecular Devices, US).
Functional analysis of hepatic spheroids and organoidsTo determine the levels of albumin secreted over time by spheroids and organoids, co-culture media were collected from perfusion channels and graft chambers (day 3, 5, and 7 for spheroids, day 7, 14, and 21 for organoids) and analyzed using the Human Albumin ELISA Quantification kit (Bethyl Laboratories Inc., E80-129, sample dilution 1:125).
Urea production was determined by a colorimetric assay kit (Biovision, #K375-100, sample dilution 1:5) following the manufacturer’s protocol.
Viability determinationFor this experiment, vascular beds were formed using HUVECs (Lonza, #2519AS) using a modified concentration of S1P (250 nM) and PMA (10 ng/ml), and endothelial cells were sprouted for 3 days. Spheroids were composed of 7500 UHH from donor 151–03. AZA was administered in both, perfusion lanes and graft chamber at a concentration range from 0.66 µM to 160 µM for 72 h (days 2–5 of co-culture with microvessels). CellTiter Glo3D (Promega, #G9681) was used to determine the toxic effect of Azathioprine on liver cultures. For this, medium was aspirated from all wells and replaced with 50 µl of a 1:1 mixture of CellTiter Glo3D reagent and Hanks' Balanced Salt Solution (HBSS, Gibco #14,025,092). The plate was then shaken for 15 min at 300 rpm (1 mm displacement) to lyse the cells and initiate the light reaction. 24 µl of the lysate was transferred to a white 384-well plate and luminescence was recorded. Luminescence was measured from lysate in graft chambers and perfusion lanes separately. Relative luminescence units were normalized to untreated condition which was defined 100% viable.
Azathioprine exposureAfter 5 days of co-culture, vascularized spheroid co-cultures were exposed to 50 µM Azathioprine (Sigma-Aldrich, PHR1282) or to vehicle control (0.1% DMSO). 50 µl of 50 µM Azathioprine or vehicle control was added to all perfusion channels in-and outlets (in MV2 medium) and to the graft chamber (in HPM medium), and the plates were placed back in the incubator on an interval rocker switching between a + 14° and -14° inclination every 8 min (OrganoFlow S, Mimetas). After 48 h, medium was collected for LDH quantification, and the perfusion assay was performed. Subsequently, medium was removed, and 5 µM of SytoX™ Green Nucleic Acid Stain (Thermo Fisher Scientific, #S7020) in MV2 was added to all perfusion channel in- and outlets and to the graft chamber for 15 min. Dead cells were visualized using a ImageXpress XLS Micro High content imaging system at 37 °C (4 × and 10 × objective). Dead nuclei quantification was performed using FIJI v.1.52 by thresholding cropped images (removing the lateral perfusion channels) using the “default” algorithm followed by “watershed” and “analyze particles.” LDH release was performed using LDH™-Glo cytotoxicity assay (Promega, #J2380, sample dilution 1:75) according to the manufacturer’s instructions.
Statistical analysisStatistical analysis was done in GraphPad Prism 8 (GraphPad Software, USA), and statistical significance was attributed to values of P < 0.05 as determined by Student’s t test or two-way ANOVA analysis, as described in the figure legends. Data are expressed as mean ± SD or SEM as described in the figure legends.
留言 (0)