
記住我

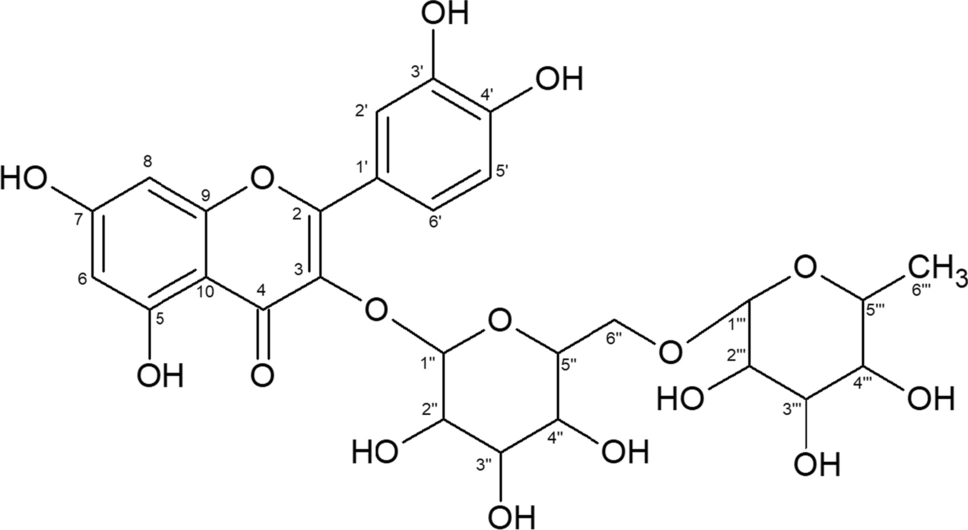

The amount of 20 µL orange peel extract was focused with cyclohexane over 10 mm separation distance. After drying the plate, a separation over 50 mm was achieved in 13 min using the solvent mixture cyclohexane–n-heptane (3:7, V/V). The dry plate was scanned with the diode-array scanner in the wavelength range from 200 to 300 nm. In Fig. 2, the result is visualized under Fig. 2A. The absorption values are plotted in colour and were calculated from the measured data according to Eq. (3).
Fig. 2
HPTLC–DAD scan of an orange peel extract (A), separated with the solvent mixture cyclohexane–n-heptane (3:7, V/V). At top, the densitogram is plotted, measured at 215 nm (B). At left is plotted the UV spectrum of ß-caryophyllene (peak No. 1) in the range of 200–300 nm (C). The data were calculated according to Eq. (3)
At top, the densitogram of the separation is plotted (Fig. 2B), measured at 215 nm (as an average of eleven diode signals). The UV spectrum of ß-caryophyllene (peak No. 1) at left (Fig. 2C) is measured in the wavelength range of 200–300 nm (spectrum averaged over nine diodes). The coloured part shows the diode-array signals of seven compounds drawn as a contour plot in the separation range from 5 to 55 mm and in the wavelength range from 200 to 300 nm. Peak 6 shows light absorption beyond 400 nm and is probably ζ-carotene [19]. Peak 6 was separated from the broad application zone only by the concentration step with cyclohexane.
HPTLC is a separation technique for non-volatile compounds because all volatile compounds evaporate when the sample is sprayed onto the plate. The compound ß-caryophyllene is the peak at 47 mm separation distance. In the system of silica gel and cyclohexane/n-heptane with its polar stationary and nonpolar mobile phase, ß-caryophyllene moves the farthest and is thus the least polar compound in the sample. When the plate is scanned several times, it can be seen that the ß-caryophyllene signal rapidly decreases in intensity. Therefore, it is not surprising that there are only a few articles on volatile compounds such as the liquids α-humulene, ß-caryophyllene [20], or thujone [21] that can be separated by HPTLC. Comparing Fig. 1 with Fig. 2, one can see that HPTLC begins where GC ends.
Polarity range B (cyclohexane–methyl tert-butyl ether, 8.6:1.4, V/V)The following separation with the solvent mixture cyclohexane–methyl tert-butyl ether (MTBE) (8.4:1.6, V/V) over 50 mm was achieved in 21 min without chamber saturation. An aliquot of 5 µL of the original extract was applied. The track was scanned and stained with vanillin reagent. The contour plot in the range from 200 to 400 nm is shown in Fig. 3A, above the stained track (bottom Fig. 3B). The data were calculated according to Eq. (3).
Fig. 3
Contour plot of an orange peel extract (A), separated with the solvent mixture cyclohexane–MTBE (8.4:1.6, V/V). In (B), the vanillin-stained track is visualized. The data were calculated according to Eq. (3)
In Fig. 3, compounds 1–7 moved unresolved near the front. In the not fully resolved multi-peak (11–14), the compounds neral and geranial have been identified with neral having the lower RF value according to [22]. Both isomers in combination are known as citral, which smells intensely of lemon. Their absorption maximum is at 250 nm, indicating a double bond in conjunction with a keto group. The compounds of peaks 9, 12 and 15 probably have a single double bond in the molecule and compounds 10 and 17 probably have two double bonds that are in conjugation. The compounds of peaks 8 and 16 show no UV absorptions. Peaks 8, 9, 12 and 16 can be stained intensely with vanillin reagent. ß-Caryophyllene (peak 1) and ζ-carotene (peak 6) can also be stained intensely, while peaks 10 and 17 show no reaction with vanillin reagent.
Figure 4 shows a separation using the same solvent system as in Fig. 3, but with 30 min chamber saturation. 10 µL of 1:5 concentrated extract was applied. In this system, the compounds of peaks 11 to 17 move to lower RF values compared to Fig. 3, because some of the ether evaporates from the plate surface during separation. That makes it difficult to compare RF values of different developments. The absolute values differ, but the pattern of the separated zones as well as the peak spectra is the same. This is what makes diode-array spectrometry in conjunction with a staining reaction so valuable for identifying peaks. Plate developing with or without chamber saturation is a tool that allows the RF values to be varied within a polarity window so that selectivity can be changed.
Fig. 4
A Contour plot with densitogram (top) recorded at 416 nm and spectrum of compound 17 (left). B VIS photograph of the track, separated with the solvent mixture cyclohexane–MTBE (8.4:1.6, V/V) without chamber saturation. C UV–VIS spectrum of compound 17 in the wavelength range 200–550 nm. The data were calculated according to Eq. (3)
Figure 4A shows the contour plot of a separation with the same solvent mixture as in Fig. 3, but separated without chamber saturation. The densitogram (top) was recorded at 416 nm. Below (Fig. 4B) the visible photograph of the track is shown. The peaks 8 and 9, 17–19 and 21 show intense yellow colours. The spectrum of compound 17 (Fig. 4C, left) shows a triplet around 400 nm typical for carotenoid [19]. Peak 12 dominates the separation with its intense orange colour. It is probably ß-cryptoxanthin (hydroxy-β-carotene) [19]. Three other carotenoids were probably separated between peaks 10 and 11, but they do not appear in the densitogram as well as peaks 13 to 16. Additional yellow bands (probably also carotenoids) are seen near the point of application.
Polarity range C (cyclohexane–methyl tert-butyl ether, 7:3, V/V)Figure 5 shows the separation over 60 mm in 19 min using the solvent mixture cyclohexane–MTBE (7:3, V/V). An aliquot of 5 µL of the original extract was applied on plate. Shown are the contour plot in the wavelength range from 200 to 500 nm (Fig. 5A) and the vanillin-stained track (Fig. 5B). The data were calculated according to Eq. (3).
Fig. 5
Contour plot in the wavelength range from 200 to 500 nm (A) and vanillin-stained track (B) of an orange peel extract, separated with the solvent mixture cyclohexane–MTBE (7:3, V/V). The data were calculated according to Eq. (3)
Geranial and neral (peaks 13 and14) are well separated. Both peaks show intense signals in the contour plot, but only weakly coloured bands after staining. Between peaks 14 and 15, there appears to be an additional zone in the contour plot that has been marked “?”. This signal does not show a coloured zone after staining and could not be detected in Fig. 3, probably due to its low amount. Therefore, signal was not counted as a peak.
The multiplet at 14 mm separation distance is shown in Fig. 3 (indicated as peaks 18–20), and in Fig. 5 a triplet at 30 mm separation distance is shown. This underlines that expression (8) is correct and the best resolution power is achieved at RF values around RF = 1/3. In Fig. 6, this multiplet again appears unresolved at 50 mm separation distance. Their absorptions beyond 400 nm identify the associated substances as carotenoids.
Fig. 6
Contour plot (A) and vanillin-stained track (B) of an orange peel extract separated with the solvent mixture cyclohexane–MTBE (3:7, V/V). The data were calculated according to Eq. (3)
Compound 23 (in the unresolved peak 23 in Fig. 5) shows a strong red zone after vanillin staining at 20 mm separation distance. This zone can be seen in Fig. 6 at 48 mm separation distance.
Polarity range D (cyclohexane–methyl tert-butyl ether, 3:7, V/V)Figure 6 shows the separation over 60 mm in 18 min using cyclohexane–MTBE (7:3, V/V). An aliquot of 5 µL of the original extract was applied. Shown are the contour plot in the wavelength range from 200 to 400 nm (Fig. 6A) and the vanillin-stained track (Fig. 6B).
In the contour plot in Fig. 6, the flavonoids naringenin (peak 25) and hesperetin (peak 26) can be identified. Both compounds show similar spectra with absorption maxima around 300 nm. In contrast, ferulic acid and p-coumaric acid (peaks 28 and 27) show different spectra with absorption maxima between 300 and 350 nm. All these compounds show only weak signals after vanillin staining. Peak 30 is the compound xanthotoxin, a coumarin derivative showing a strong fluorescence under UV 366 nm irradiation.
Below a separation distance of 20 mm, very intense signals can be seen which all have similar UV spectra and all show deep yellow-coloured zones after vanillin staining.
Polarity range E (methyl tert-butyl ether)Figure 7 shows the separation over 60 mm in 19 min using MTBE as solvent. An aliquot of 0.5 µL of the original extract was applied. The contour plot in the range from 200 to 400 nm (Fig. 7A) is plotted below the densitogram (Fig. 7B), taken at 359 nm. The UV absorption spectrum of sinensetin (Fig. 7C) is shown on the left.
Fig. 7
Contour plot of an orange peel extract (A) separated with MTBE solvent. The data were analysed according to Eq. (3). At the top is plotted the densitogram (B), measured at 359 nm. At left is plotted the UV spectrum of sinensetin (compound No. 36) in the range from 200 to 400 nm (C)
Peak 30 shows strong fluorescence and could be identified as xanthotoxin. Compared to its absorbance intensity, at least six compounds (peaks 31–36) are separated, showing much higher intensity. These group of compounds comprises by far the most abundant substances in the orange peel extract. From the mass spectra of peaks 35 and 36, it appears that these compounds are nobiletin and sinensetin, a fivefold and a sixfold methoxylated flavone, respectively. This suggests that peaks 34 and 33 are compounds 3,5,6,7,3′,4′-hexamethoxyflavone and 3',4',5,5',6,7-hexamethoxyflavone, respectively.
In orange peel juice, TLC revealed the compounds sinensetin (5,6,7,3',4'-pentamethoxyflavone), nobiletin (5,6,7,8,3',4'-hexamethoxyflavone), 3,5,6,7,8,3',4'-heptamethoxyflavone, tetra-O-methylscutellarein (5,6,7,4'-tetramethoxyflavone) and tangeretin (5,6,7,8,4'-pentamethoxyflavone), separated with increasing RF values in that order [23]. In the literature, RP-HPLC separations of orange peel extracts have reported 4 to 6 compounds that have high concentrations and elute with sinensetin first [24,25,26,27]. These are the compounds sinensetin, 3',4',5,5',6,7-hexamethoxyflavone, nobiletin and tangeretin [24], sinensetin, nobiletin, tangeretin and two unidentified compounds [25], nobiletin, tangeretin, sinensetin, 5,6,7,4′-tetramethoxyflavone, 3,5,6,7,3′,4′-hexamethoxyflavone, and 3,5,6,7,8,3′,4′-heptamethoxyflavone [26], and sinensetin, nobiletin, 3,5,6,7,8,3′,4′-heptamethoxyflavone, and tangeretin [27]. In commercial orange juices, the compounds sinensetin, 3',4',5,5',6,7-hexamethoxyflavone, nobiletin, heptamethoxyflavone, tetra-O-methylscutellarein, and tangeretin were determined [28].
All this agrees well with our own result and suggests that peak 32 in Fig. 7 should be tangeretin. However, from the literature review, the question arises as to which compound is separated as peak 31. The DART–TOF–MS spectrum (Fig. 8) of this zone shows two strong signals at 433 m/e and 449 m/e. The signal at 433 m/e indicates a molecule in which two C–H groups of tangeretin are replaced by two –O–CH3 groups. This is probably the compound 3,3',4',5,6,7,8-heptamethoxyflavone. The signal at 449 m/e could have an additional C–OH group instead of a C–H group. The DART–TOF–MS spectrum in Fig. 8 is a clear indication that peak 31 is not pure.
Fig. 8
DART–TOF–MS of peak 31. The signals at m/e 433 and 449 are interesting
It is also a strange result that the six absorption peaks in Fig. 7 show only four strong fluorescence signals, with sinensetin showing by far the most intense fluorescence. The first two fluorescence peaks correlate well with peaks 35 and 36, but the next two fluorescence signals are problematic. The third signal lies in between the RF values of peaks 33 and 34, and the fourth fluorescence signal has its maximum at the left slope of signal 31. All this indicates that peaks 31 to 33 in Fig. 7 are not pure.
Figure 9 shows the fluorescence signals of a separation over 60 mm in 18 min using MTBE–CH2Cl2 (3.5:6.5, V/V) as solvent. Five different fluorescence signals can be seen. In Fig. 9A, the contour plot of the fluorescence signals is plotted, calculated according to Eq. (4). In Fig. 9B, the track of a tenfold higher amount obtained under UV 366 nm illumination is plotted. The densitogram is shown above, measured at 365 nm (Fig. 9C). At left, the fluorescence spectrum of sinensetin (peak No. 36) is plotted in the range from 400 to 700 nm (Fig. 9D). For illumination to generate fluorescence, a diode is used which emits light at 365 nm. The diode emits a much higher intensity of light than the detector can resolve, and thus the detector renders a saturated signal. In other words, J and J0 have the same value. If the spectral data are evaluated according to Eq. (4), the LED signals will be rendered as zero. This can be seen in spectrum D of Fig. 9.
Fig. 9
Fluorescence contour plot of an orange peel extract separated with the solvent mixture MTBE–CH2Cl2 (3.5:6.5, V/V). Data were calculated according to Eq. (4). A Shown is the contour plot of the fluorescence signals. B The track of a tenfold higher sample amount, illuminated with UV 366 nm light. At the top is plotted the densitogram, measured at 365 nm (C). At left, the fluorescence spectrum of sinensetin (peak No. 36) is plotted in the range from 400 to 700 nm (D)
The fluorescence peak 34 in Fig. 9 fits well with the absorption peak of 34 in Fig. 7. The fluorescence peak 33 fits well with the absorption peak 32 in Fig. 7, and the fluorescence peak 31 fits well with the absorption peak of 31 in Fig. 7. According to the literature, tangeretin (peak 32) shows a weak yellow fluorescence [23], but this is obviously suppressed by the strong blue fluorescence of peak 33. It follows that the peak at 52 mm separation distance in Fig. 9 must contain compounds 32 and 33 and is therefore not pure. By HPTLC, we find at least six compounds with high concentration in the extract separated near sinensetin (peaks 31 to 36).
In the fluorescence contour plot of Fig. 9A, peak 31 shows a symmetrical shape and a blue fluorescence. Interestingly, a yellow fluorescent zone can be observed at higher concentrations when the plate is illuminated with UV 366 nm light. The contour plot in Fig. 9A was measured on a track where 0.5 µL extract was separated. The track shown in Fig. 9B is a separation of 5 µL of extract. The tenfold amount of zone 31 shows a small yellow band on the right side of its blue zone that cannot be from tangeretin. The corresponding contour plot signal (31a) shows at least three peaks, with the fluorescence of the right region shifted to higher wavelengths (over 575 nm). This is a second indication that peak 31 contains different compounds.
Two-dimensional separation in the polarity range EThe polarity window shown in Fig. 9 (polarity range E) clearly contains more compounds than HPTLC can separate. In such a case, HPTLC opens the possibility to separate in a second dimension. The plate only needs to be dried and developed perpendicular to the first direction with a second solvent, which should preferably have orthogonal properties. Roughly calculated, we have separated eleven fluorescent zones in Fig. 9B. Assuming round peaks of diameter d and a separation number of SN = 11, the maximum capacity for a two-dimensional separation is 154 peaks, calculated according to Eq. (9).
$$}\left( \right) = \frac}^$$
(9)
Figure 10 shows a two-dimensional separation of 0.5 µL orange peel extract on a 10 × 10 cm silica gel 60 HPTLC plate. In the first direction, the plate was developed with cyclohexane–MTBE (3:7, V/V) over 60 mm and then with MTBE–CH2Cl2 (6.5:3.5, V/V) in 16 min. For development in the second direction over 60 mm in 13 min, the solvent mixture toluene–ethyl acetate–methanol (55:45:5, V/V) was used. At left and top, the standards sinensetin, nobiletin and tangeretin were separated in addition to 0.5 µL of extract each. The sinensetin standard shows three side peaks (top), nobiletin shows two side peaks (best seen at left) and the weak yellow fluorescent tangeretin shows a single contamination.
Fig. 10
A Fluorescence evaluation at 366 nm illumination of a two-dimensional separation. In the first direction, the plate was developed using cyclohexane–MTBE (3:7, V/V) and after drying was developed again with the solvent mixture MTBE–CH2Cl2 (3.5:6.5, V/V). In the second direction, the plate was developed with the solvent mixture toluene–MTBE–methanol (55:45:5, V/V). B After staining with vanillin, the plate was evaluated under UV 366 nm light
In Fig. 10, it can be seen that only spot 34 is pure, as it has no side peaks. Sinensetin (peak 36) shows two side peaks (36a and 36b), peak 33 three additional peaks (33a–c), and peak 31 shows three contaminations (31a–c). In the orange peel extract, a tangeretin (peak 32) cannot be detected under UV 366 nm because its amount is below the detection limit. Of interest is the nobiletin spot (peak 35), which appears to have only a single contaminant, detected as 35a under UV 366 illumination (Fig. 10A), but staining with vanillin (shown in Fig. 10B) reveals a second side peak (35b). In Fig. 10A, a series of fluorescent spots can be seen, starting with peak 37. In summary, there are many more methoxylated flavones present in an orange peel extract than have detected by HPLC and HPTLC to date. Preparative chemistry has known this for a long time [29].
Polarity range F (ethyl acetate–ethanol, 9:1, V/V)Figure 11 shows the separation of the original orange peel extract as a fluorescence contour plot, separated over 60 mm in 14 min using ethyl acetate–ethanol (9:1, V/V) as solvent mixture. In Fig. 11A, the fluorescence densitogram of the separation is shown, measured at 450 nm. In Fig. 11B, the contour plot of the fluorescence signals is plotted, which was evaluated according to Eq. (4). In Fig. 11C, the unstained track is shown, illuminated with UV 366 nm light. In Fig. 11D, the track is shown after staining with vanillin reagent and measured under visible light. In Fig. 11E, the fluorescence spectrum of peak 38 is plotted, measured in the wavelength range of 400–700 nm.
Fig. 11
Fluorescence contour plot of an orange peel extract, separated with the solvent mixture ethyl acetate–ethanol (9:1, V/V). Data were evaluated according to Eq. (4). A Shown is the fluorescence densitogram measured at 450 nm. B Contour plot of the fluorescence signals. C Track of the separation taken under UV 366 nm. D The track stained with vanillin reagent and measured under VIS light. E: The fluorescence spectrum of peak 38 in the wavelength range of 400–700 nm is shown at left
In Fig. 11C, at least ten fluorescent zones can be distinguished. The two strong zones 37 and 38 are followed by two weaker zones (39 and 40), which show only two small peaks in the densitogram (Fig. 11A). Peak No. 42 is rather broad and probably consists of three unresolved peaks. In Fig. 11D, some yellow zones can be seen. These yellow zones are probably from methylated flavones. Peaks 41 and 43 will also belong to this group of compounds.
Polarity range G (ethyl acetate–ethanol–formic acid, 8.8:1:0.2, V/V)Figure 12 shows the separation of 5 µL of the original orange peel extract as fluorescence contour plot over 60 mm in 15 min using the solvent mixture ethyl acetate–ethanol–formic acid (8.8:1:0.2, V/V). After drying, the plate was stored in NH3 vapour for 2 min to neutralize the formic acid. The raw data were evaluated according to Eq. (5), the original Kubelka–Munk equation. Using this equation for spectral evaluation often results in sharper peaks [15]. In Fig. 12A, the contour plot of the absorption signals is plotted, measured in the wavelength range 200–500 nm. In Fig. 12B, the separation track of a fourfold sample amount is shown, stained with vanillin reagent. The densitogram is plotted above, measured at 343 nm (Fig. 12C), and the absorption spectrum of peak 46 is plotted on the left in the wavelength range of 200–500 nm (Fig. 12D).
Fig. 12
Kubelka–Munk contour plot of an orange peel extract, separated with the solvent mixture ethyl acetate–ethanol–formic acid (8.8:1:0.2, V/V). The data were calculated according to Eq. (5). A Shown is the contour plot of the Kubelka–Munk signals, measured in the wavelength range 200–500 nm. B Separation track of a fourfold more sample amount, stained with vanillin reagent. C Kubelka–Munk densitogram measured at 343 nm. D The Kubelka–Munk spectrum of a carotenoid (peak No. 46) is plotted in the range of 200–500 nm
In Fig. 12A, peak No. 46 dominates. In Fig. 12D, the Kubelka–Munk spectrum of this carotenoid is plotted in the wavelength range 200–500 nm. Only four peaks are registered between this peak and the application point (at a separation distance of 5 mm). No compound is remaining at the point of application, so the separation of polarity range G is the last of the GMD sequence. It is interesting to note that neither naringin nor hesperidin has been identified so far. Both compounds are flavanone glycosides with RF values in the polarity range G of 0.27 and 0.05, respectively. Both compounds are too polar to dissolve in the upper phase of the ethyl acetate/water extract. To separate and detect all polar compounds of the aqueous phase, a second GMD sequence must be performed.
留言 (0)